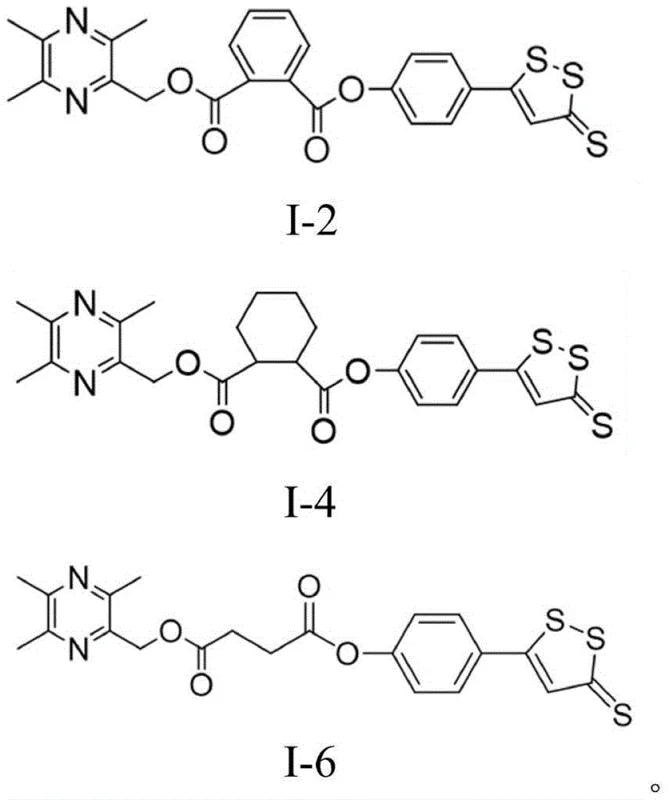
Advanced Synthesis of Potent Ligustrazine Derivatives for Cardiovascular Drug Development
The pharmaceutical landscape for cardiovascular therapeutics is continuously evolving, driven by the urgent need for more potent anti-platelet agents with improved safety profiles. Patent CN111825664B, published in early 2023, introduces a significant breakthrough in this domain through the development of novel ligustrazine derivatives, specifically compounds designated as I-2, I-4, and I-6. These molecules represent a strategic structural modification of the natural product tetramethylpyrazine (TMP), aiming to overcome the limitations of the parent compound such as weak physiological activity and poor water solubility. The disclosed technology provides a robust synthetic pathway that not only enhances biological efficacy but also offers a viable route for the manufacturing of high-purity pharmaceutical intermediates. For R&D teams and procurement specialists seeking reliable sources for next-generation cardiovascular drug candidates, understanding the chemical architecture and synthetic feasibility of these derivatives is paramount. The structural diversity achieved in this patent allows for targeted modulation of platelet aggregation pathways, positioning these compounds as valuable assets in the pipeline for treating ischemic heart disease and stroke.
In the context of modern medicinal chemistry, the transition from a lead compound to a clinically viable drug candidate often hinges on the ability to efficiently synthesize analogues with optimized pharmacokinetic properties. Conventional methods for modifying tetramethylpyrazine have historically struggled with regioselectivity and yield, often requiring harsh conditions that degrade the sensitive pyrazine ring or the sulfur-containing side chains. Traditional approaches might involve direct alkylation which can lead to complex mixtures of isomers, necessitating difficult and costly separation processes that hinder commercial viability. Furthermore, the integration of the 1,2-dithiole-3-thione moiety, known for its cytoprotective effects, onto the pyrazine scaffold has been chemically challenging due to compatibility issues between the reactive functional groups. These legacy limitations result in extended development timelines and inflated costs for raw material acquisition, creating bottlenecks for supply chain managers who require consistent quality and volume.
The novel approach detailed in the patent circumvents these historical hurdles through a convergent synthetic strategy that modularly assembles the target molecules. As illustrated in the comprehensive reaction scheme, the process begins with the independent preparation of two key fragments: the phenolic dithiole-thione derivative (ADTOH) and the hydroxymethyl-tetramethylpyrazine intermediate. This divergence allows for the optimization of each fragment's synthesis before the final coupling, thereby minimizing impurity carryover and maximizing overall throughput. The core innovation lies in the use of a mild esterification protocol to link these fragments, avoiding the high temperatures and aggressive reagents that typically compromise yield in similar heterocyclic systems. By decoupling the synthesis of the sulfur-rich domain from the nitrogen-rich pyrazine core until the final stages, the method ensures that the delicate 1,2-dithiole ring remains intact, preserving the biological activity essential for the drug's mechanism of action. This strategic design significantly simplifies the purification workflow, making it an attractive option for cost reduction in pharmaceutical intermediate manufacturing.
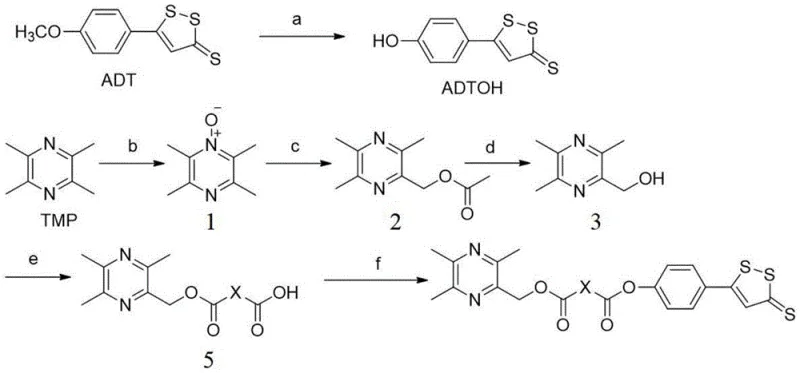
Mechanistically, the synthesis relies heavily on the efficiency of the Steglich esterification and related acylation reactions mediated by carbodiimides. The transformation of tetramethylpyrazine (TMP) into the key alcohol intermediate involves a controlled oxidation sequence using hydrogen peroxide in acetic acid, followed by acetylation and hydrolysis. This sequence is critical for installing the reactive hydroxymethyl handle at the 2-position of the pyrazine ring without over-oxidizing the methyl groups or the ring nitrogens. Subsequently, this alcohol reacts with various dianhydrides (phthalic, hexahydrophthalic, or succinic anhydride) in the presence of 4-dimethylaminopyridine (DMAP). The DMAP acts as a potent nucleophilic catalyst, accelerating the formation of the mono-ester intermediate while suppressing the formation of di-esters or other side products. The final coupling with ADTOH utilizes dicyclohexylcarbodiimide (DCC) to activate the remaining carboxylic acid group, forming an O-acylisourea intermediate that is rapidly attacked by the phenolic hydroxyl of ADTOH. This mechanism is particularly advantageous for impurity control, as the byproduct, dicyclohexylurea (DCU), is largely insoluble in many organic solvents and can be removed via simple filtration, thereby streamlining the downstream processing and ensuring the high purity required for regulatory compliance.
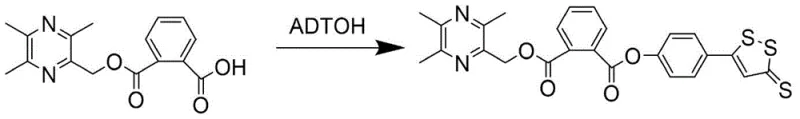
To further illustrate the precision required in executing this synthesis, consider the specific coupling reaction used to generate compound I-2. The reaction between the pyrazine-phthalic acid mono-ester and ADTOH must be carefully monitored to prevent racemization or degradation of the dithiole ring. The use of dichloromethane as a solvent provides an optimal medium for solubility while maintaining a temperature profile that favors the desired ester linkage. TLC monitoring is employed to determine the exact endpoint, ensuring that the expensive ADTOH starting material is fully consumed without prolonged exposure to the coupling reagents which could lead to decomposition. This level of procedural control is indicative of a mature process capable of being transferred from the laboratory bench to pilot plant reactors, providing confidence to stakeholders regarding the reproducibility of the chemical identity and potency of the final active pharmaceutical ingredient.
How to Synthesize Ligustrazine Derivatives Efficiently
The synthesis of these high-value cardiovascular intermediates requires strict adherence to the optimized reaction parameters outlined in the patent to ensure maximum yield and purity. The process involves distinct stages including the demethylation of ADT, the oxidation of TMP, and the final convergent coupling, each demanding specific attention to temperature, stoichiometry, and workup procedures. For research and development teams aiming to replicate or scale this chemistry, understanding the nuances of the purification steps—such as the specific solvent ratios for column chromatography described in the examples—is crucial for isolating the target isomers from potential regioisomers. The detailed standardized synthesis steps provided below serve as a foundational guide for laboratory execution, though industrial scaling may require adaptation of purification techniques from chromatography to crystallization for economic feasibility.
- Preparation of ADTOH via demethylation of ADT using pyridine hydrochloride at high temperature.
- Oxidation of Tetramethylpyrazine (TMP) to form the intermediate alcohol (3,5,6-trimethylpyrazin-2-yl)methanol.
- Coupling of the pyrazine alcohol with various dianhydrides and subsequent esterification with ADTOH using DCC/DMAP catalysts.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the synthetic route disclosed in CN111825664B offers substantial advantages that directly address the pain points of procurement managers and supply chain directors in the pharmaceutical industry. The reliance on readily available starting materials such as tetramethylpyrazine and anethole dithiole thione (ADT) ensures a stable supply base, mitigating the risks associated with sourcing exotic or single-source reagents. Furthermore, the modular nature of the synthesis allows for the parallel production of intermediates, which can drastically reduce the overall lead time for high-purity pharmaceutical intermediates. By avoiding the use of precious metal catalysts or cryogenic conditions, the process inherently lowers the operational expenditure associated with energy consumption and specialized equipment maintenance. This translates into a more competitive cost structure for the final API, enabling pharmaceutical companies to price their cardiovascular therapies more aggressively in the global market while maintaining healthy margins.
- Cost Reduction in Manufacturing: The elimination of transition metal catalysts and the use of ambient temperature conditions for the critical coupling steps significantly reduce the utility costs and waste treatment expenses associated with heavy metal removal. The high atom economy of the esterification reaction minimizes raw material waste, and the ability to filter off solid byproducts like DCU simplifies the workup, reducing solvent usage and processing time. These factors collectively contribute to a leaner manufacturing process that drives down the cost of goods sold (COGS) without compromising on the quality or potency of the therapeutic agent.
- Enhanced Supply Chain Reliability: The synthetic pathway utilizes commodity chemicals that are widely produced and traded in the global fine chemical market, ensuring that supply disruptions are unlikely to halt production. The robustness of the reaction conditions, which tolerate minor variations in temperature and mixing rates, means that the process can be reliably executed across different manufacturing sites or by contract manufacturing organizations (CMOs). This flexibility empowers supply chain heads to diversify their vendor base and secure long-term contracts with reliable pharmaceutical intermediate suppliers, ensuring continuity of supply for critical cardiovascular medications.
- Scalability and Environmental Compliance: The process is designed with scalability in mind, utilizing standard unit operations such as reflux, extraction, and filtration that are easily adaptable from kilogram to ton-scale production. The absence of highly toxic reagents or persistent organic pollutants aligns with increasingly stringent environmental regulations, reducing the burden of waste disposal and permitting. This environmental compatibility not only safeguards the manufacturer against regulatory fines but also enhances the corporate sustainability profile, which is becoming a key criterion for partnership selection in the modern pharmaceutical supply chain.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these novel ligustrazine derivatives. These insights are derived directly from the experimental data and claims presented in the patent documentation, providing clarity on the feasibility and performance of the technology. Understanding these aspects is essential for stakeholders evaluating the potential integration of these intermediates into their drug development pipelines or procurement strategies.
Q: What is the primary pharmacological advantage of Ligustrazine derivatives I-2, I-4, and I-6 over the parent compound TMP?
A: According to patent CN111825664B, these derivatives exhibit significantly superior inhibitory activity against ADP-induced and AA-induced platelet aggregation compared to the parent tetramethylpyrazine (TMP), with efficacy comparable to or exceeding standard drugs like ticlopidine and aspirin.
Q: What are the key reaction conditions for the final esterification step?
A: The final coupling reaction utilizes dichloromethane as the solvent with DCC (dicyclohexylcarbodiimide) and DMAP (4-dimethylaminopyridine) as catalysts, typically proceeding at room temperature to ensure high yield and purity.
Q: Is this synthesis route scalable for industrial production?
A: Yes, the route relies on standard organic unit operations such as reflux, extraction, and crystallization. While the patent describes column chromatography for lab-scale purification, industrial processes can optimize this via recrystallization to ensure cost-effectiveness and scalability.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Ligustrazine Derivative Supplier
As the demand for advanced cardiovascular therapeutics grows, the need for a partner who can bridge the gap between innovative patent chemistry and commercial reality becomes critical. NINGBO INNO PHARMCHEM stands at the forefront of this intersection, leveraging extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our state-of-the-art facilities are equipped to handle the specific requirements of heterocyclic chemistry, ensuring that the stringent purity specifications demanded by global regulatory bodies are consistently met. With rigorous QC labs and a deep understanding of the nuances involved in synthesizing sulfur-nitrogen containing scaffolds, we are uniquely positioned to support your transition from clinical trials to full-scale market launch.
We invite you to engage with our technical procurement team to discuss how we can tailor our manufacturing capabilities to your specific project needs. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into how our optimized processes can reduce your overall development budget. We encourage potential partners to contact us directly to obtain specific COA data and route feasibility assessments, ensuring that your supply chain is built on a foundation of scientific excellence and commercial reliability.