Optimizing Exatecan Intermediate Production: A High-Yield Seven-Step Synthetic Strategy for Global Pharma Supply Chains
Optimizing Exatecan Intermediate Production: A High-Yield Seven-Step Synthetic Strategy for Global Pharma Supply Chains
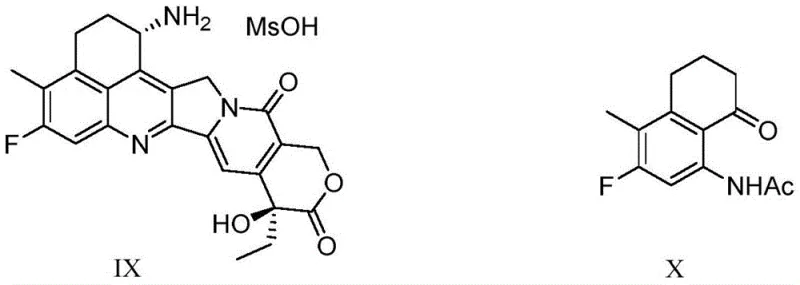
The global demand for potent anticancer agents continues to drive innovation in the synthesis of complex pharmaceutical intermediates, particularly for topoisomerase I inhibitors like Exatecan (DX-8951). As a critical synthetic analog of camptothecin, Exatecan requires high-purity precursors to ensure clinical efficacy and safety in treating advanced solid tumors. Recent advancements detailed in patent CN114516808A, published on May 20, 2022, introduce a groundbreaking preparation method for amino-protected benzocyclic ketone compounds, which serve as the pivotal building blocks for Exatecan. This technology addresses long-standing inefficiencies in the supply chain by offering a robust, seven-step synthetic route that significantly outperforms historical methodologies in both yield and operational simplicity. For procurement leaders and R&D directors seeking a reliable pharmaceutical intermediate supplier, understanding the mechanistic advantages of this new pathway is essential for securing a stable supply of high-value oncology ingredients.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of the key intermediate, Compound X, has been plagued by low efficiency and hazardous processing conditions that hinder commercial scalability. Early attempts, such as those disclosed in patent JP 3-015812, relied on a seven-step sequence starting from 2-fluoro-toluene but suffered from severe safety risks due to the requirement for low-temperature borane reductions and high-temperature polyphosphoric acid reactions. More critically, the cyclization step using polyphosphoric acid yielded a dismal 25%, creating a massive bottleneck in material throughput. Subsequent efforts, including the eleven-step route in WO96/26181, attempted to resolve these issues but resulted in a cumulative total yield of merely 17% due to excessive Friedel-Crafts acylations and complex carbonyl reduction sequences. Even more recent approaches, like the eight-step Heck coupling route in WO2019/044946, struggled with low yields during nitration and halogenation steps, capping the total process efficiency at approximately 14%. These legacy methods not only inflate production costs through material loss but also complicate purification due to the formation of isomeric by-products.
The Novel Approach
In stark contrast to these inefficient legacy pathways, the methodology described in CN114516808A utilizes a strategic seven-step sequence that leverages modern cross-coupling chemistry to achieve a total yield exceeding 40%. The core innovation lies in the early introduction of the side chain via a Negishi coupling reaction, which bypasses the need for multiple acylation and reduction steps found in older routes. Furthermore, the process employs a clever regiocontrol strategy where a bromine atom is introduced to direct the subsequent nitration, ensuring precise placement of the amino functionality without generating difficult-to-remove isomers. This streamlined approach not only drastically reduces the number of unit operations but also utilizes milder reaction conditions that are inherently safer for commercial scale-up of complex pharmaceutical intermediates. By eliminating the reliance on hazardous polyphosphoric acid cyclizations and improving atom economy, this novel route represents a paradigm shift in the manufacturing of acetamidotetralone derivatives.
Mechanistic Insights into Negishi Coupling and Regioselective Functionalization
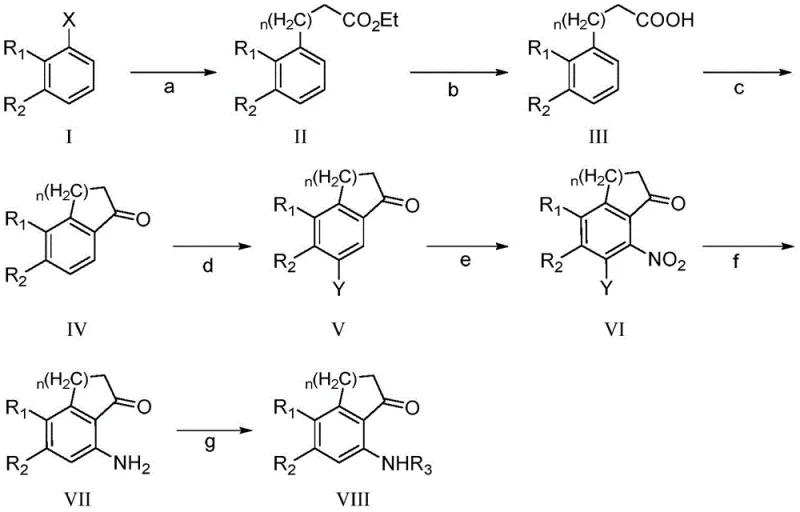
The success of this synthetic strategy hinges on the precise execution of the initial Negishi coupling (Step a), where a halogenated aromatic precursor reacts with an organozinc reagent to install the requisite carbon chain. This transformation is catalyzed by palladium species, such as palladium acetate, in conjunction with bulky phosphine ligands like S-PHOS or X-PHOS, which facilitate the oxidative addition and reductive elimination cycles necessary for high conversion. Operating at moderate temperatures between 20°C and 60°C in anhydrous THF or DMF, this step avoids the extreme conditions often required for traditional Grignard additions, thereby preserving sensitive functional groups on the aromatic ring. Following side-chain installation, the intramolecular Friedel-Crafts acylation (Step c) closes the ring to form the tetralone core. Unlike prior art methods that struggle with regioselectivity, this protocol utilizes protic acids like trifluoroacetic acid combined with dehydrating agents to drive the cyclization efficiently at low temperatures (-10°C to 5°C), minimizing polymerization and tar formation.
Further downstream, the introduction of the nitrogen functionality is achieved through a highly controlled sequence of bromination and nitration. The presence of the bromine substituent acts as a powerful ortho/para director, guiding the nitro group to the exact position required for the final amine, a critical factor in maintaining high purity standards. The subsequent hydrogenation (Step f) reduces the nitro group to an amine while simultaneously removing the bromine directing group if necessary, or retaining it depending on the specific derivative target. Crucially, the addition of organic bases like triethylamine during hydrogenation neutralizes the hydrogen halide by-products generated in situ, preventing the formation of ammonium salts that could complicate isolation. This meticulous attention to mechanistic detail ensures that the final amino-protected product is obtained with minimal impurity profiles, satisfying the rigorous quality specifications demanded by high-purity pharmaceutical intermediate markets.
How to Synthesize Amino-protected Benzocyclic Ketone Efficiently
The practical implementation of this synthesis involves a logical progression of well-established organic transformations adapted for industrial robustness. Starting from commercially available halogenated toluenes, the process builds molecular complexity through carbon-carbon bond formation followed by ring closure and functional group manipulation. Each step has been optimized to maximize yield and minimize workup complexity, such as the direct use of crude products from the coupling step in subsequent hydrolysis without intermediate purification. This telescoping capability is vital for reducing solvent consumption and processing time in a manufacturing setting. For technical teams looking to implement this chemistry, the detailed standardized synthetic steps are outlined below to ensure reproducibility and safety compliance.
- Perform Negishi coupling on a halogenated toluene derivative with an organozinc reagent to introduce the side chain.
- Execute intramolecular Friedel-Crafts acylation under acid catalysis to form the benzocyclic ketone core.
- Conduct regioselective bromination followed by nitration, then reduce the nitro group and protect the amine.
Commercial Advantages for Procurement and Supply Chain Teams
From a strategic sourcing perspective, the adoption of this seven-step synthesis offers profound benefits for cost reduction in API manufacturing by fundamentally altering the economics of intermediate production. The most immediate impact is the dramatic improvement in overall process yield, which jumps from the historical average of 14-25% to over 40%. In chemical manufacturing, such a doubling of efficiency directly translates to a substantial reduction in the cost of goods sold (COGS), as less raw material is wasted and fewer batches are required to meet production targets. Additionally, the elimination of hazardous reagents like borane and polyphosphoric acid reduces the need for specialized containment equipment and expensive waste disposal protocols, further lowering the operational overhead. The use of readily available starting materials, such as 2-bromo-6-fluorotoluene, ensures that the supply chain remains resilient against raw material shortages, providing procurement managers with greater negotiating power and supply security.
- Cost Reduction in Manufacturing: The streamlined seven-step route eliminates the need for multiple redundant acylation and reduction cycles found in eleven-step legacy processes, significantly cutting down on reagent consumption and energy usage. By avoiding low-yielding steps like the polyphosphoric acid cyclization which previously offered only 25% yield, the new method maximizes material throughput and minimizes the volume of organic waste generated per kilogram of product. This efficiency gain allows manufacturers to offer more competitive pricing structures while maintaining healthy margins, making it an attractive option for generic drug developers seeking to optimize their bill of materials.
- Enhanced Supply Chain Reliability: The reliance on robust, scalable reactions such as Negishi coupling and catalytic hydrogenation ensures that the production process is less susceptible to the variability often seen in exotic or highly sensitive chemistries. Since the raw materials are commodity chemicals rather than custom-synthesized precursors, lead times for sourcing inputs are significantly shortened, facilitating faster response to market demand fluctuations. Furthermore, the simplified purification procedures, which avoid difficult chromatographic separations of isomers, enable faster batch release times, thereby reducing lead time for high-purity pharmaceutical intermediates reaching the final formulation stage.
- Scalability and Environmental Compliance: The reaction conditions specified in the patent, such as operating temperatures between 0°C and 60°C, are easily manageable in standard stainless steel reactors without the need for cryogenic cooling or high-pressure autoclaves. This compatibility with existing infrastructure lowers the barrier to entry for contract manufacturing organizations (CMOs) looking to adopt the technology. Moreover, the improved atom economy and reduced solvent intensity align with modern green chemistry principles, helping pharmaceutical companies meet their sustainability goals and regulatory environmental standards without compromising on output quality or volume.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this novel synthesis pathway. These insights are derived directly from the experimental data and comparative analysis provided in the patent documentation, offering clarity on how this method resolves specific pain points associated with Exatecan intermediate production. Understanding these details is crucial for technical evaluators assessing the feasibility of technology transfer.
Q: What is the primary advantage of this new synthesis route over prior art?
A: The primary advantage is a significant increase in total yield to over 40%, compared to 14-25% in previous methods, achieved by shortening the route to seven steps and improving regioselectivity during nitration.
Q: How does the process ensure high regioselectivity for the amino group placement?
A: The method utilizes a strategic bromine atom as a directing group prior to nitration, which ensures accurate positioning of the nitro group and subsequent amine, avoiding the formation of difficult-to-separate isomers common in older routes.
Q: Is this synthesis method suitable for large-scale industrial manufacturing?
A: Yes, the process uses readily available raw materials like 2-bromo-6-fluorotoluene and employs standard reaction types such as Negishi coupling and hydrogenation, making it highly scalable and safe for commercial production without extreme low-temperature hazards.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Amino-protected Benzocyclic Ketone Supplier
As the pharmaceutical industry continues to evolve, the ability to access advanced synthetic technologies is a key differentiator for maintaining a competitive edge in the oncology sector. NINGBO INNO PHARMCHEM stands at the forefront of this evolution, leveraging deep expertise in process chemistry to deliver complex intermediates with unmatched consistency and quality. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet the rigorous demands of global supply chains. With our stringent purity specifications and rigorous QC labs, we guarantee that every batch of amino-protected benzocyclic ketone meets the highest international standards, providing our partners with the confidence needed to advance their clinical and commercial programs.
We invite procurement specialists and R&D leaders to engage with us for a Customized Cost-Saving Analysis tailored to your specific project requirements. By partnering with our technical procurement team, you can gain access to specific COA data and comprehensive route feasibility assessments that demonstrate the tangible economic benefits of switching to this optimized synthesis. Contact us today to discuss how our advanced manufacturing capabilities can support your long-term supply strategy and drive value across your organization.