Advanced Synthetic Route for High-Purity Olmesartan Intermediate via Optimized Grignard Reaction
Introduction to Patent CN102060778A Technology
The pharmaceutical industry continuously seeks robust synthetic pathways for critical cardiovascular intermediates, specifically for the production of Angiotensin II receptor blockers like Olmesartan. Patent CN102060778A, published on May 18, 2011, discloses a highly efficient method for synthesizing ethyl 4-(1-hydroxyl-1-methylethyl)-2-propyl imidazole-5-carboxylate, a pivotal building block in this therapeutic class. This technology represents a significant leap forward in process chemistry, addressing long-standing issues regarding yield loss and impurity profiles associated with traditional 2-substituted 4,5-dinitrile imidazole synthesis. By leveraging a streamlined four-step sequence that integrates imidate formation, cyclization, direct alcoholysis, and Grignard addition, the inventors have achieved a remarkable total yield of 81%, drastically outperforming the historical benchmark of 55%. For R&D directors and procurement specialists, this patent offers a blueprint for cost reduction in API manufacturing through enhanced atom economy and simplified unit operations.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 2-propyl-imidazole derivatives has been plagued by inefficient solvent management and difficult purification challenges. Conventional literature methods typically utilize acetonitrile for the initial condensation of diaminomaleonitrile with orthoesters, followed by a solvent swap to methanol or xylene for the cyclization step. This multi-solvent approach not only increases operational complexity but also leads to significant material loss during distillation and transfer. Furthermore, prior art processes often suffer from the formation of polar, black viscous substances during the cyclization phase, which are notoriously difficult to remove. The standard practice of adding activated carbon post-cyclization frequently fails to adequately decolorize the reaction mixture, resulting in poor crystal quality and compromised purity. These technical bottlenecks severely limit the commercial scale-up of complex pharmaceutical intermediates, forcing manufacturers to accept lower yields and higher production costs.
The Novel Approach
In stark contrast, the methodology outlined in CN102060778A introduces a cohesive strategy that optimizes both chemical efficiency and physical handling. The novel route maintains a consistent alcoholic solvent system (methanol or ethanol) throughout the initial imidate formation and the subsequent ammonolysis, effectively eliminating the need for intermediate solvent recovery. A critical innovation is the strategic timing of the decolorization step; activated carbon is introduced immediately after the imidate formation and prior to the ammonia-induced cyclization. This proactive purification prevents the entrapment of colored impurities within the crystal lattice of the dinitrile intermediate. Additionally, the process merges the hydrolysis and esterification of the nitrile groups into a single direct alcoholysis step using hydrogen chloride in ethanol. This consolidation reduces the number of isolation steps and minimizes exposure to harsh conditions that could degrade the sensitive imidazole core.
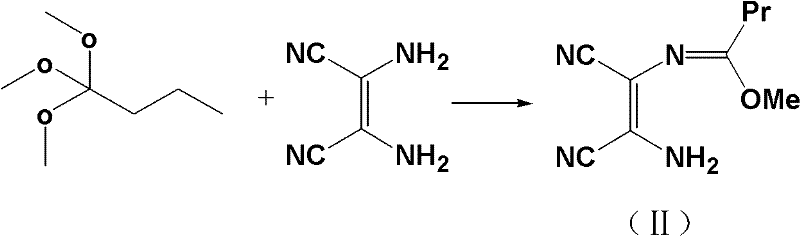
Mechanistic Insights into Direct Alcoholysis and Grignard Addition
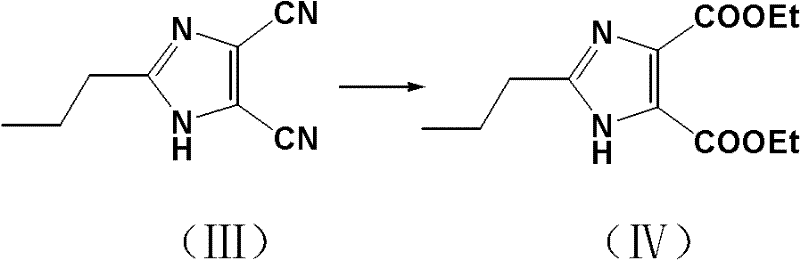
The chemical elegance of this synthesis lies in the mechanism of the direct alcoholysis of the dinitrile intermediate. Traditionally, converting nitriles to esters involves a two-stage process: acid-catalyzed hydrolysis to the carboxylic acid followed by Fischer esterification. However, this patent utilizes a Pinner-like reaction variant where the dinitrile is refluxed directly in an ethanol solution containing hydrogen chloride. In this acidic alcoholic medium, the nitrile nitrogen is protonated, increasing the electrophilicity of the cyano carbon, which is then attacked by the ethanol nucleophile to form an imidate salt intermediate. Subsequent hydrolysis of this imidate yields the ethyl ester directly. This one-pot transformation is thermodynamically favorable and kinetically efficient, bypassing the isolation of the unstable dicarboxylic acid. The reaction conditions are finely tuned, with the patent specifying that higher concentrations of hydrogen chloride (up to 40%) can reduce reaction times from 7 days to just 2 days, demonstrating excellent process controllability.
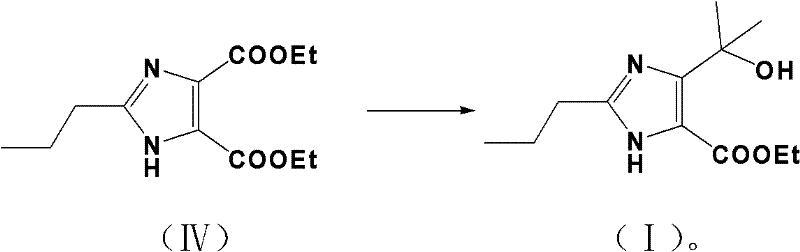
Following the formation of the diethyl ester, the final transformation involves a highly selective Grignard reaction to install the tertiary alcohol moiety. The reaction of diethyl 2-propylimidazole-4,5-dicarboxylate with methylmagnesium bromide is a delicate operation requiring precise stoichiometric control. The mechanism involves the nucleophilic attack of the methyl carbanion on the carbonyl carbon of the ester at the 4-position. To ensure complete conversion to the tertiary alcohol without stopping at the ketone stage, a significant excess of the Grignard reagent (5:1 molar ratio) is employed. The reaction is conducted at low temperatures (0-5°C) in anhydrous THF or ether under an inert nitrogen atmosphere to prevent side reactions such as enolization or attack on the remaining ester at the 5-position, although the electronic environment of the imidazole ring generally favors selective mono-addition under these controlled conditions. The result is the formation of the target 4-(1-hydroxyl-1-methylethyl) group with high regioselectivity and purity exceeding 99%.
How to Synthesize Ethyl 4-(1-hydroxyl-1-methylethyl)-2-propyl Imidazole-5-carboxylate Efficiently
Implementing this synthesis requires strict adherence to the optimized parameters defined in the patent to replicate the high yields and purity levels. The process begins with the preparation of the imidate intermediate, followed by immediate purification and cyclization, setting the stage for high-quality downstream processing. The subsequent direct alcoholysis and Grignard steps demand careful monitoring of temperature and reagent addition rates to manage exotherms and ensure safety. For detailed operational protocols, equipment specifications, and safety guidelines necessary for GMP-compliant manufacturing, please refer to the standardized synthesis guide below.
- Reflux trimethyl orthobutyrate and diaminomaleonitrile in alcohol solvent, add activated carbon for decolorization, and filter hot.
- Cool filtrate to 0-15°C, pass ammonia gas to cyclize and form 2-propylimidazole-4,5-dicarbonitrile.
- React the dinitrile intermediate with hydrogen chloride in ethanol under reflux to perform simultaneous hydrolysis and esterification.
- Treat the resulting diethyl ester with methylmagnesium bromide (Grignard reagent) in THF or ether to generate the final tertiary alcohol product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of the technology described in CN102060778A offers tangible strategic benefits beyond mere technical superiority. The shift from a 55% yield process to an 81% yield process fundamentally alters the cost structure of the intermediate. Higher yields mean less raw material consumption per kilogram of finished product, directly impacting the variable cost of goods sold. Furthermore, the simplification of the workflow—specifically the elimination of solvent swaps and the merging of hydrolysis/esterification steps—reduces the overall cycle time and utility consumption (steam, cooling water, electricity). These efficiencies translate into a more competitive pricing model for the final API, allowing pharmaceutical companies to maintain healthy margins even in price-sensitive markets.
- Cost Reduction in Manufacturing: The most significant economic driver of this new method is the dramatic improvement in overall yield. By increasing the total yield from 55% to 81%, the process effectively reduces the theoretical consumption of starting materials like diaminomaleonitrile and trimethyl orthobutyrate by nearly one-third. Additionally, the elimination of the solvent exchange step between imidate formation and cyclization removes the capital and operational expenses associated with large-scale distillation and solvent recovery systems. The use of common, low-cost solvents like ethanol and methanol throughout the sequence further minimizes procurement costs compared to specialized solvents like acetonitrile or xylene. This streamlined approach ensures that the cost reduction in pharmaceutical intermediate manufacturing is realized through both material efficiency and operational simplicity.
- Enhanced Supply Chain Reliability: Supply chain continuity is often threatened by complex processes that are prone to failure or batch rejection. The improved decolorization strategy, where activated carbon is used prior to crystallization, consistently produces white crystalline intermediates with high purity (99.8% for the dinitrile and 99.7% for the diester). High-purity intermediates reduce the risk of downstream failures during the final API synthesis, thereby stabilizing the supply chain. Moreover, the robustness of the direct alcoholysis step, which tolerates a range of reaction times (2 to 7 days) depending on acid concentration, provides manufacturing flexibility. This flexibility allows production planners to adjust batch schedules without compromising product quality, ensuring reducing lead time for high-purity intermediates becomes a manageable operational goal rather than a bottleneck.
- Scalability and Environmental Compliance: From an environmental and scalability perspective, this route is superior. The reduction in the number of unit operations inherently lowers the volume of waste solvent generated. By avoiding the use of benzene or xylene, which are often subject to stricter environmental regulations due to toxicity concerns, the facility reduces its regulatory burden. The use of ethanol and methanol facilitates easier waste treatment and potential solvent recycling. The high purity of the final product (99.2%) minimizes the need for extensive recrystallization or chromatographic purification at the API stage, further reducing the environmental footprint. This alignment with green chemistry principles makes the process highly scalable and attractive for facilities aiming to meet rigorous international environmental standards while expanding production capacity.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis route. These answers are derived directly from the experimental data and beneficial effects reported in patent CN102060778A, providing clarity on process capabilities and limitations. Understanding these details is crucial for technical teams evaluating the feasibility of technology transfer.
Q: How does this patent improve upon conventional synthesis methods for Olmesartan intermediates?
A: The patent CN102060778A improves the total yield from the conventional 55% to 81% by merging the hydrolysis and esterification steps into a single direct alcoholysis reaction and optimizing the decolorization timing to occur before cyclization, preventing the formation of black viscous byproducts.
Q: What are the critical process parameters for the Grignard reaction step?
A: The Grignard reaction requires strict temperature control between 0-5°C using methylmagnesium bromide in THF or ether under nitrogen protection, with a molar ratio of reagent to substrate of 5:1 to ensure complete conversion to the tertiary alcohol without side reactions.
Q: Why is the solvent consistency in steps 1 and 2 significant for industrial production?
A: Using the same low-bo-point alcoholic solvent (methanol or ethanol) for both the imidate formation and the subsequent ammonolysis eliminates the need for solvent exchange or distillation between steps, significantly reducing energy consumption and processing time while minimizing waste generation.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Ethyl 4-(1-hydroxyl-1-methylethyl)-2-propyl Imidazole-5-carboxylate Supplier
At NINGBO INNO PHARMCHEM, we recognize that the successful commercialization of cardiovascular therapeutics depends on the availability of high-quality, cost-effective intermediates. Our technical team has extensively analyzed the pathway described in CN102060778A and possesses the expertise to execute this synthesis with precision. We bring extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to industrial reactor is seamless. Our facilities are equipped with stringent purity specifications and rigorous QC labs capable of verifying the 99.2% purity benchmark required for this critical Olmesartan intermediate, guaranteeing that every batch meets the exacting standards of global regulatory bodies.
We invite potential partners to engage with our technical procurement team to discuss how this optimized synthetic route can be integrated into your supply chain. By collaborating with us, you gain access to a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to request specific COA data and route feasibility assessments to validate the performance metrics discussed in this report. Let us help you secure a reliable supply of this essential pharmaceutical intermediate while maximizing your operational efficiency.