Advanced Solid-Phase Synthesis of Teixobactin Analogs for Commercial Antibiotic Production
The pharmaceutical landscape is constantly evolving in the search for next-generation antibiotics that can overcome multidrug resistance, and patent CN106632604B represents a significant breakthrough in this domain by disclosing a robust preparation method for Teixobactin analogs. Discovered originally through iChip technology, native Teixobactin showed immense promise due to its unique mechanism of destroying bacterial cell walls without inducing resistance; however, its natural isolation was plagued by low yields and complex processing. This patent addresses those critical bottlenecks by introducing a streamlined solid-phase synthesis strategy that utilizes simple, cost-effective amino acid raw materials to construct complex macrocyclic peptide scaffolds. By shifting from reliance on uncultured soil bacteria to a controlled chemical synthesis environment, this technology enables the rapid generation of structural variants essential for comprehensive pharmacodynamic evaluation and future drug development. The method specifically leverages hydrazine resin or similar solid supports to facilitate efficient chain elongation and subsequent on-resin modifications, marking a pivotal step towards the commercial viability of this novel antibiotic class.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior to this innovation, the acquisition of Teixobactin and its derivatives was hindered by severe technical and logistical constraints inherent in both natural extraction and early synthetic attempts. The original isolation method using iChip technology, while groundbreaking for discovery, is fundamentally unsuitable for industrial scale-up due to its reliance on the in-situ culture of unculturable bacteria, a process that is time-consuming, labor-intensive, and yields insufficient quantities for extensive clinical testing. Furthermore, early synthetic approaches described in prior art often relied on liquid-phase synthesis systems or complex solid-phase strategies involving orthogonal protecting groups like Alloc (allyloxycarbonyl). These conventional routes necessitated numerous synthesis steps, intricate deprotection sequences, and the handling of sensitive intermediates in solution, which collectively resulted in low overall yields and prohibitive costs. The involvement of liquid-phase cyclization steps in particular introduced purification challenges and scalability issues that prevented the rapid exploration of structure-activity relationships (SAR) needed to optimize the drug candidate.
The Novel Approach
In stark contrast, the methodology outlined in CN106632604B offers a simplified and highly efficient pathway that bypasses the complexities of previous techniques by utilizing a specialized solid-phase strategy. This novel approach involves the sequential coupling of 6 to 12 amino acids directly onto a functionalized resin, followed by a unique on-resin esterification step that builds the cyclic precursor without requiring premature cleavage. By selecting specific amino acids with hydroxyl side chains, such as Serine or Threonine, as branching points, the synthesis creates a pre-organized linear peptide that is primed for macrocyclization. This strategy eliminates the need for cumbersome Alloc protecting groups and reduces the total number of synthetic operations, thereby enhancing the overall throughput. The result is a versatile platform capable of generating a diverse library of analogs, such as Arg10-teixobactin and Lys10-teixobactin, with high purity and consistency, effectively transforming a difficult-to-access natural product into a manufacturable pharmaceutical intermediate.
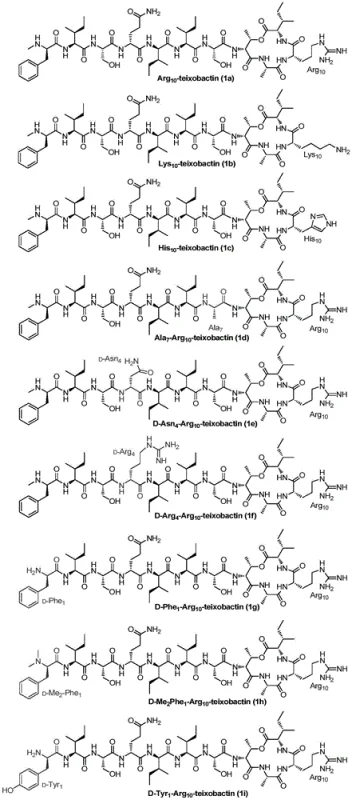
Mechanistic Insights into Copper-Mediated Macrocyclization
The core chemical innovation driving the success of this synthesis lies in the precise orchestration of peptide bond formation and macrocyclization under mild conditions. The process begins with the assembly of a linear peptide chain on a solid support, where the penultimate residue is selected for its hydroxyl functionality to serve as a nucleophile for subsequent ester bond formation. Once the linear sequence is established, two additional amino acids are condensed onto this side chain hydroxyl group, creating a branched precursor that contains the necessary components for ring closure. The critical cyclization step is then achieved not through traditional high-dilution solution methods, but by cleaving the peptide from the resin and subjecting it to a copper acetate and pyridine system. This copper-mediated oxidation facilitates the formation of the amide bond between the terminal amino group and the carboxyl group, effectively closing the macrocycle with high efficiency. The use of copper acetate is particularly advantageous as it promotes the reaction under relatively mild conditions, minimizing the risk of epimerization or degradation of the sensitive peptide backbone, which is a common pitfall in the synthesis of large cyclic peptides.
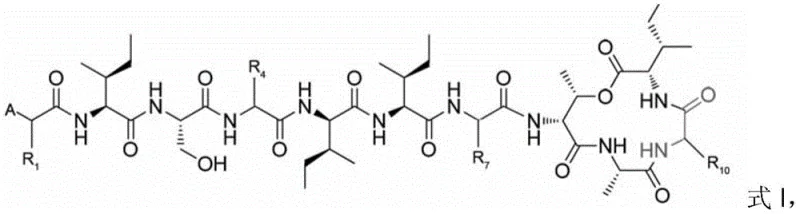
Furthermore, the control of impurities and stereochemistry is rigorously maintained throughout the synthesis through the use of standard Fmoc chemistry and careful monitoring of coupling efficiency. The protocol incorporates detection methods such as the chloranil test or ninhydrin assay at each coupling stage to ensure complete reaction before proceeding, which is vital for preventing the accumulation of deletion sequences that are difficult to separate later. The final deprotection step utilizes a trifluoroacetic acid (TFA) based cocktail, typically comprising TFA, triisopropylsilane (TIS), and water in a specific ratio, to cleanly remove acid-labile side-chain protecting groups without compromising the integrity of the newly formed macrocycle. This meticulous attention to reaction conditions ensures that the final Teixobactin analogs possess the correct stereochemistry and high chemical purity required for biological activity, distinguishing this method from less controlled liquid-phase alternatives that often struggle with racemization and byproduct formation.
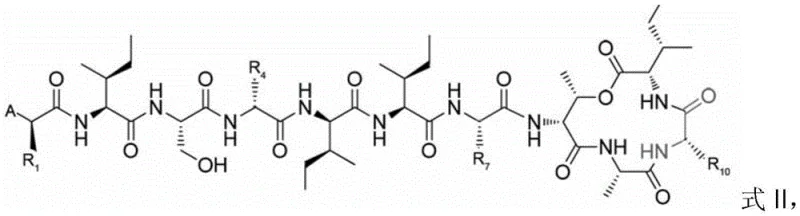
How to Synthesize Teixobactin Analogs Efficiently
The synthesis of these high-value antibiotic intermediates follows a logical progression of solid-phase assembly, precursor modification, and solution-phase cyclization that can be adapted for various scales of production. The detailed standardized synthesis steps involve specific reagents and conditions optimized to maximize yield and minimize waste, ensuring that the process is robust enough for technology transfer. For a comprehensive understanding of the exact molar ratios, solvent volumes, and reaction times required to replicate this success, please refer to the structured guide below which outlines the critical operational parameters derived from the patent examples.
- Sequentially couple 6-12 amino acids onto a solid-phase resin with amino or hydrazine groups to form a linear peptide, ensuring the penultimate residue contains a hydroxyl side chain.
- Perform condensation coupling of two additional amino acids onto the side chain hydroxyl group of the penultimate residue to form an ester-linked precursor.
- Cleave the peptide from the resin and induce macrocyclization by coupling the terminal amino group with the carboxyl group using a copper acetate method.
- Remove side-chain protecting groups using a TFA-based cleavage cocktail, purify via semi-preparative HPLC, and lyophilize to obtain the final analog.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthetic route offers transformative benefits that directly address the cost and availability challenges associated with novel antibiotics. By replacing the unpredictable supply chain of natural extraction with a deterministic chemical synthesis process, manufacturers can secure a stable and continuous supply of Teixobactin analogs essential for clinical trials and eventual commercialization. The reliance on simple, commodity-grade amino acids as starting materials drastically reduces the raw material cost base compared to methods requiring exotic or custom-synthesized building blocks. Moreover, the elimination of complex protecting group strategies like Alloc simplifies the inventory management of reagents and reduces the consumption of specialized catalysts, leading to significant operational cost savings. This streamlined workflow also shortens the overall production cycle time, allowing for faster response to market demands and reducing the working capital tied up in long manufacturing campaigns.
- Cost Reduction in Manufacturing: The economic viability of this process is underpinned by the use of inexpensive, widely available amino acid feedstocks and the avoidance of costly orthogonal protecting groups. By simplifying the synthesis to a more linear solid-phase protocol with fewer deprotection and purification steps, the overall consumption of solvents and reagents is significantly lowered. This reduction in process complexity translates directly into lower manufacturing costs per gram of active pharmaceutical ingredient, making the development of Teixobactin-based drugs more financially attractive for pharmaceutical partners.
- Enhanced Supply Chain Reliability: Unlike natural product isolation which is subject to biological variability and environmental factors, this chemical synthesis method offers a highly reliable and reproducible supply source. The ability to synthesize the compound on demand using standard peptide synthesis equipment mitigates the risk of supply disruptions and ensures consistent quality across different batches. This reliability is crucial for maintaining the continuity of drug development programs and meeting regulatory requirements for material consistency during the transition from preclinical to clinical stages.
- Scalability and Environmental Compliance: The solid-phase nature of the initial synthesis steps allows for easy scalability from milligram research quantities to kilogram pilot production without fundamental changes to the chemistry. Furthermore, the process generates waste streams that are typical of peptide synthesis and can be managed using standard industrial effluent treatment protocols. The high efficiency of the copper-mediated cyclization also minimizes the formation of byproducts, reducing the burden on downstream purification and waste disposal systems, thereby aligning with modern green chemistry principles and environmental regulations.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these Teixobactin analogs, providing clarity on the advantages of this specific patent technology. These insights are derived directly from the experimental data and background analysis presented in the patent documentation, offering a transparent view of the method's capabilities and limitations for potential partners and stakeholders.
Q: How does this synthetic method improve upon natural extraction via iChip technology?
A: Unlike the iChip technology which requires complex in-situ culture and separation of uncultured soil bacteria, this solid-phase synthesis method uses simple, commercially available amino acids. It significantly reduces production time and complexity, allowing for rapid development of derivatives and pharmacodynamic evaluation without the bottlenecks of natural product isolation.
Q: What is the key advantage of the cyclization strategy described in patent CN106632604B?
A: The method utilizes a copper acetate/pyridine system for cyclization after cleaving the linear peptide from the resin. This avoids the need for complex protecting group strategies like Alloc often found in prior art, simplifying the synthesis workflow and reducing the number of deprotection steps required to achieve the macrocyclic structure.
Q: Which bacterial strains are targeted by these Teixobactin analogs?
A: The synthesized analogs, such as Arg10-teixobactin and Lys10-teixobactin, demonstrate potent inhibitory activity against Gram-positive bacteria, including Staphylococcus aureus and Bacillus subtilis. They are designed to disrupt bacterial cell wall synthesis without inducing resistance, offering a promising alternative to traditional antibiotics.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Teixobactin Analog Supplier
As the global demand for effective antibiotics continues to rise, NINGBO INNO PHARMCHEM stands ready to support your development goals with our advanced capabilities in peptide synthesis and process optimization. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet the rigorous volume requirements of late-stage clinical trials and market launch. Our state-of-the-art facilities are equipped with stringent purity specifications and rigorous QC labs to guarantee that every batch of Teixobactin analog meets the highest international standards for safety and efficacy. We understand the critical nature of antibiotic supply chains and are committed to delivering high-quality intermediates with the consistency and reliability that top-tier pharmaceutical companies demand.
We invite you to collaborate with us to leverage this cutting-edge synthesis technology for your antibiotic pipeline. Please contact our technical procurement team to request a Customized Cost-Saving Analysis tailored to your specific volume needs. We are prepared to provide specific COA data and route feasibility assessments to demonstrate how our manufacturing expertise can accelerate your project timelines and optimize your overall development costs.