Scalable Synthesis of Barnidipine Hydrochloride: A Technical Breakthrough for API Manufacturing
Scalable Synthesis of Barnidipine Hydrochloride: A Technical Breakthrough for API Manufacturing
The global demand for effective antihypertensive agents continues to drive innovation in the synthesis of dihydropyridine calcium channel blockers. Among these, Barnidipine Hydrochloride (CAS: 104757-53-1) stands out as a potent, long-acting therapeutic agent with high vascular selectivity. The efficient production of this complex molecule relies heavily on robust synthetic methodologies that ensure high optical purity and economic viability. Patent CN101643469A introduces a refined seven-step synthesis pathway that addresses historical bottlenecks in manufacturing, specifically focusing on the strategic timing of chiral resolution and the elimination of chromatographic purification. This technical insight report analyzes the proprietary reaction sequence, highlighting its potential to redefine supply chain stability for pharmaceutical manufacturers seeking a reliable pharmaceutical intermediate supplier.
The structural complexity of Barnidipine Hydrochloride necessitates precise control over stereochemistry, particularly at the C4 position of the dihydropyridine ring and the chiral center of the pyrrolidine side chain. As illustrated in the molecular structure below, the integration of the 3-nitrophenyl group and the specific stereochemical configuration are critical for biological activity. The patented process achieves this through a convergent strategy that builds the dihydropyridine core first, resolves the chirality at the mono-acid stage, and finally couples the chiral side chain. This approach minimizes the loss of expensive chiral reagents and streamlines the downstream processing requirements.
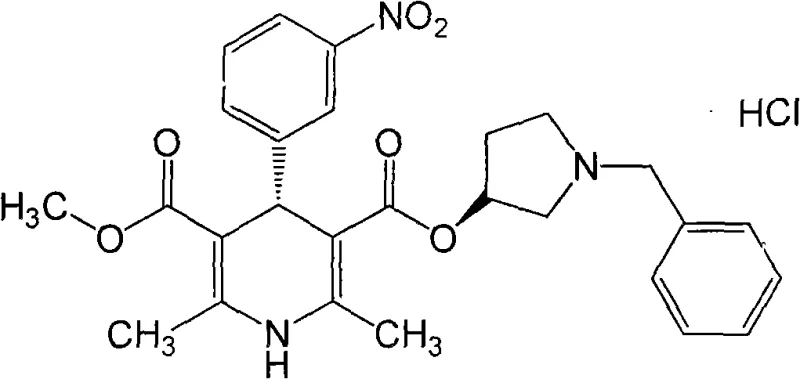
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of Barnidipine Hydrochloride has been plagued by inefficiencies that hinder large-scale production. Prior art methods often relied on the synthesis of racemic mixtures followed by late-stage resolution, a strategy that inherently caps the maximum theoretical yield at 50% unless dynamic kinetic resolution is employed. Furthermore, earlier techniques frequently utilized tert-butyl esters or required extensive purification via silica gel column chromatography to remove diastereomeric impurities. These chromatographic steps are notoriously difficult to scale, consuming vast quantities of solvents and generating significant hazardous waste, which directly contradicts modern green chemistry principles and inflates the cost of goods sold (COGS). Additionally, some legacy routes involved one-pot reactions that resulted in complex impurity profiles, making it challenging to meet the stringent purity specifications required for active pharmaceutical ingredients (APIs).
The Novel Approach
The methodology outlined in CN101643469A represents a paradigm shift by introducing an early-stage resolution strategy coupled with a column-free purification protocol. Instead of resolving the final bulky ester, the process targets the monocarboxylic acid intermediate (Compound IX) for chiral separation using organic bases such as cinchonidine. This tactical decision ensures that the expensive chiral alcohol, 1-benzyl-3-pyrrolidinol, is only reacted with the desired enantiomer, drastically reducing raw material costs. Moreover, the entire process is designed to rely on crystallization and liquid-liquid extraction for purification. By eliminating the need for column chromatography, the process becomes inherently more scalable, safer, and environmentally compliant, offering a distinct competitive advantage for cost reduction in API manufacturing.
Mechanistic Insights into Chiral Resolution and Hantzsch Cyclization
The core of this synthesis lies in the construction of the 1,4-dihydropyridine ring via a modified Hantzsch synthesis, followed by a critical differentiation of the ester groups. The reaction begins with the condensation of 3-hydroxypropionitrile and diketene to form an acetoacetate derivative, which then undergoes a Knoevenagel condensation with m-nitrobenzaldehyde. This intermediate subsequently reacts with ethyl aminocrotonate to close the dihydropyridine ring, yielding the symmetric diester (Compound VIII). The mechanistic elegance of the subsequent step involves the selective hydrolysis of one ester group using a strong base. This selectivity is crucial; it transforms the symmetric diester into a chiral mono-acid (upon resolution), creating a handle for stereochemical separation without disturbing the sensitive nitro group or the dihydropyridine core.
Following hydrolysis, the racemic mono-acid (Compound IX) is subjected to resolution using a chiral organic base. The patent specifies the use of alkaloids like cinchonidine or quinine, which form diastereomeric salts with the enantiomers of the acid. Due to differences in solubility, one diastereomeric salt crystallizes preferentially, allowing for physical separation. This step is the gatekeeper for optical purity. Once the pure chiral acid (Compound X) is isolated and liberated, it is activated (typically via acid chloride formation using phosphorus pentachloride) and coupled with 1-benzyl-3-pyrrolidinol. The comprehensive reaction scheme below details this entire flow, from simple nitriles to the final hydrochloride salt, demonstrating the logical progression of functional group transformations.
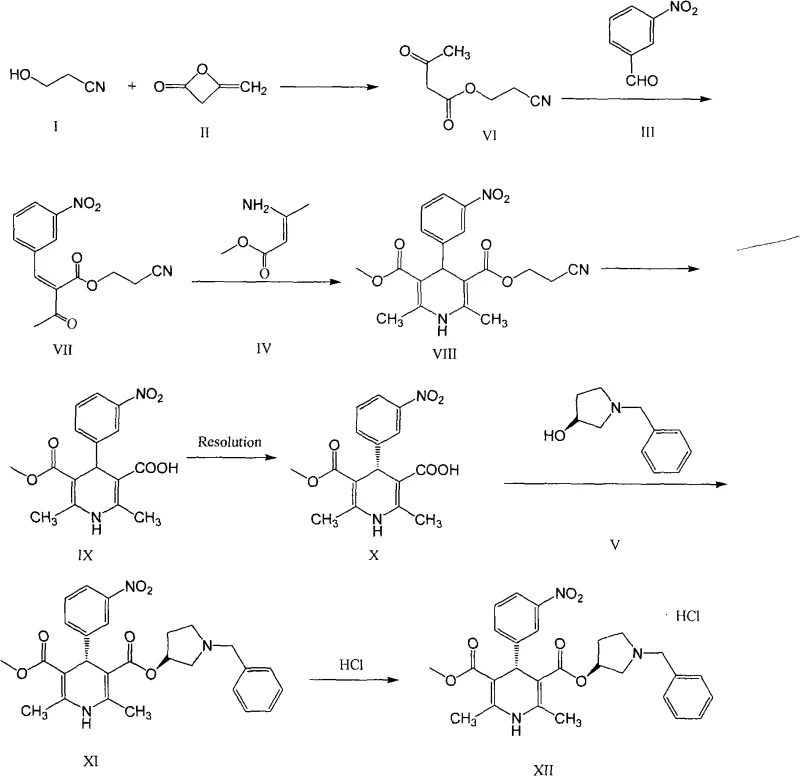
How to Synthesize Barnidipine Hydrochloride Efficiently
Executing this synthesis requires careful control of reaction parameters, particularly temperature and stoichiometry, to maximize yield and minimize byproduct formation. The process is divided into distinct operational units: precursor synthesis, core cyclization, chiral resolution, and final coupling. Each step has been optimized in the patent examples to proceed under relatively mild conditions, typically between 0°C and 100°C, utilizing common industrial solvents such as methanol, ethanol, and dichloromethane. The following guide outlines the standardized operational framework derived from the patent data, serving as a baseline for process engineers looking to implement this technology.
- Condensation of 3-hydroxypropionitrile with diketene to form the acetoacetate derivative.
- Knoevenagel condensation with m-nitrobenzaldehyde followed by Hantzsch cyclization with ethyl aminocrotonate.
- Selective hydrolysis of the diester to a mono-acid, followed by chiral resolution using an organic base like cinchonidine.
- Final esterification with 1-benzyl-3-pyrrolidinol and salt formation with hydrogen chloride.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the transition to this novel synthesis route offers tangible benefits that extend beyond mere chemical yield. The primary value driver is the substantial reduction in processing time and solvent consumption achieved by removing column chromatography. In a commercial setting, chromatography is a bottleneck that limits throughput and requires specialized equipment; its removal allows for the use of standard stainless steel reactors and centrifuges, thereby increasing batch frequency and overall plant capacity. Furthermore, the strategy of resolving the intermediate acid rather than the final ester means that the costly chiral alcohol is not wasted on the unwanted enantiomer, leading to significant raw material savings.
- Cost Reduction in Manufacturing: The elimination of silica gel column chromatography is a major cost-saving factor. Chromatography requires large volumes of high-purity solvents and generates significant waste disposal costs. By replacing this with crystallization and extraction, the process drastically lowers variable costs per kilogram. Additionally, the early-stage resolution prevents the financial loss associated with coupling expensive chiral alcohols to the wrong enantiomer, optimizing the atom economy of the most valuable reagents in the synthesis.
- Enhanced Supply Chain Reliability: The raw materials specified in this route, such as 3-hydroxypropionitrile, diketene, and m-nitrobenzaldehyde, are commodity chemicals with stable global supply chains. Unlike specialized catalysts or rare reagents, these inputs are readily available from multiple vendors, reducing the risk of supply disruption. The robustness of the reaction conditions, which tolerate a range of temperatures and do not require ultra-low temperature cryogenics for most steps, further ensures consistent production schedules regardless of seasonal or logistical variations.
- Scalability and Environmental Compliance: The process is explicitly designed for industrial suitability, avoiding laboratory-only techniques. The reliance on phase separations and crystallizations makes the technology easily transferable from pilot plants to multi-ton production scales. From an environmental perspective, the reduction in solvent usage and the avoidance of silica waste align with increasingly strict environmental regulations, simplifying the permitting process for manufacturing sites and reducing the carbon footprint of the API production lifecycle.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production of Barnidipine Hydrochloride using this specific patented methodology. These answers are derived directly from the experimental data and claims within CN101643469A, providing clarity on the feasibility and advantages of the process for potential partners and technical stakeholders.
Q: Why is chiral resolution performed on the mono-acid intermediate rather than the final ester?
A: Resolving the mono-acid intermediate (Compound X) is significantly more cost-effective because it avoids wasting the expensive chiral alcohol (benzylpyrrolol) on the unwanted enantiomer. It also simplifies purification before the final coupling step.
Q: Does this synthesis process require column chromatography?
A: No, a key advantage of this patented process (CN101643469A) is that it eliminates the need for silica gel column chromatography, relying instead on crystallization and extraction, which makes it highly suitable for industrial scale-up.
Q: What are the critical quality attributes for Barnidipine Hydrochloride intermediates?
A: Critical attributes include high optical purity (ee value) of the resolved mono-acid, control of the diastereomeric ratio during esterification, and strict limits on residual solvents and heavy metals to meet API standards.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Barnidipine Hydrochloride Supplier
At NINGBO INNO PHARMCHEM, we understand that the successful commercialization of antihypertensive APIs depends on a partner who can bridge the gap between laboratory innovation and industrial reality. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of this patent are fully realized in a GMP-compliant environment. We maintain stringent purity specifications and operate rigorous QC labs equipped to monitor optical rotation and impurity profiles at every stage, guaranteeing that the final Barnidipine Hydrochloride meets the highest global pharmacopeial standards.
We invite pharmaceutical companies and generic drug manufacturers to collaborate with us to leverage this cost-effective synthesis route. By partnering with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements. Contact us today to discuss route feasibility assessments and to obtain specific COA data for our pilot batches, ensuring your supply chain is built on a foundation of chemical excellence and reliability.