Revolutionizing Pyridino-Azepine Synthesis: A Scalable Route for High-Purity Pharmaceutical Intermediates
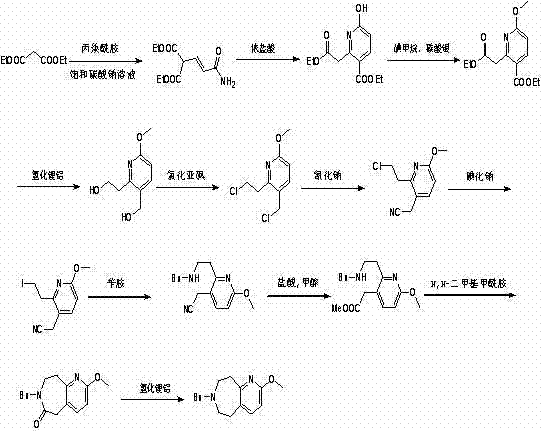
The pharmaceutical industry is constantly seeking robust synthetic pathways that balance chemical complexity with manufacturing feasibility, and patent CN103130799A presents a significant breakthrough in the synthesis of 2-hydroxy pyridino-6,7,8,9-tetrahydro-5H-pyridine[2,3-d] azepine derivatives. This specific class of heterocyclic compounds serves as a critical scaffold in the development of novel therapeutic agents, yet their historical production has been plagued by inefficient multi-step sequences and the use of hazardous reagents. The disclosed invention fundamentally re-engineers the synthetic logic by replacing a cumbersome eleven-step protocol with a concise, high-yielding route that begins with readily available propionitrile and specialized keto esters. By shifting the paradigm from sensitive propiolamide chemistry to a more stable nitrile-based addition, the patent addresses the longstanding pain points of low overall yield and difficult reaction control that have hindered the widespread adoption of this valuable chemical motif. For R&D directors and process chemists, this represents a tangible opportunity to accelerate drug discovery timelines by securing a more reliable source of high-quality intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art methodologies, such as those described in WO2009079765, relied heavily on the condensation of propiolamide with diethyl malonate, a strategy that inherently introduced significant operational risks and inefficiencies into the manufacturing workflow. The reliance on propiolamide is particularly problematic because this reagent is known for its high sensitivity and instability, requiring stringent handling conditions that complicate experimental operations and increase the likelihood of batch-to-batch variability. Furthermore, the conventional route necessitates an exhaustive eleven-step sequence to reach the target pyridino-azepine core, which inevitably leads to a dramatic erosion of overall yield as material is lost at each isolation and purification stage. This elongated process not only inflates the cost of goods sold due to excessive solvent and reagent consumption but also creates a complex impurity profile that is difficult to characterize and control, posing severe challenges for regulatory compliance in pharmaceutical manufacturing. Consequently, the industry has faced a bottleneck where the demand for these bioactive scaffolds outstrips the capacity of legacy synthesis methods to deliver them economically and safely.
The Novel Approach
In stark contrast to the convoluted legacy processes, the innovative method detailed in CN103130799A streamlines the synthesis by utilizing propionitrile and a specific keto ester as the foundational building blocks, catalyzed by potassium carbonate in an acetonitrile solvent system. This approach eliminates the need for hypersensitive starting materials, allowing the initial addition reaction to proceed smoothly from 0°C to room temperature, thereby drastically simplifying the thermal management requirements of the process. Following the formation of the adduct, the protocol employs a direct reflux in concentrated hydrochloric acid to effect ring closure, a robust transformation that tolerates scale-up far better than the delicate steps found in previous art. The final purification involves the strategic use of di-tert-butyl dicarbonate, which not only protects sensitive functionalities but also aids in the crystallization and isolation of the pure product. This condensed workflow reduces the number of unit operations, minimizes waste generation, and significantly enhances the safety profile of the entire manufacturing campaign.
Mechanistic Insights into Base-Catalyzed Nucleophilic Addition and Cyclization
The core chemical transformation driving this synthesis is a base-mediated nucleophilic addition where the methylene group of the keto ester attacks the electron-deficient triple bond of the propionitrile. In the presence of potassium carbonate, the acidic protons alpha to the carbonyl group of the keto ester are deprotonated to generate a reactive enolate species, which then undergoes a Michael-type addition to the nitrile functionality. This step is critical because the choice of acetonitrile as the solvent ensures excellent solubility for both the organic substrates and the inorganic base, facilitating efficient mass transfer and reaction kinetics without the need for exotic phase-transfer catalysts. The reaction temperature is carefully ramped from 0°C to room temperature to control the exotherm and prevent potential polymerization of the nitrile, ensuring that the adduct forms with high regioselectivity. This mechanistic precision is what allows the process to avoid the formation of tarry byproducts that often plague nitrile chemistry under harsher conditions.
Following the addition, the subsequent cyclization step in concentrated hydrochloric acid is driven by the protonation of the nitrile nitrogen, which activates the carbon atom towards intramolecular nucleophilic attack by the neighboring amine or enol functionality generated in situ. This acid-catalyzed ring closure is thermodynamically favorable and proceeds to completion under reflux conditions, effectively locking in the pyridino-azepine skeleton. The use of strong mineral acid also serves a dual purpose by hydrolyzing any remaining ester groups or protecting groups that might interfere with the final structure, effectively cleaning up the reaction mixture during the cyclization event itself. From an impurity control perspective, this one-pot cyclization and hydrolysis strategy minimizes the exposure of intermediates to air and moisture between steps, thereby reducing the risk of oxidation or hydrolysis side reactions that could compromise the purity of the final API intermediate.
How to Synthesize 2-hydroxy pyridino-azepine derivatives Efficiently
The practical execution of this synthesis requires careful attention to stoichiometry and workup procedures to maximize the recovery of the valuable heterocyclic product. The process begins with the dissolution of the keto ester in acetonitrile, followed by the slow addition of the base and the nitrile to manage heat evolution, ensuring a safe and controlled reaction environment. After the initial addition is complete and the mixture has stirred overnight to ensure full conversion, the reaction is quenched into ice water and extracted with ethyl acetate to isolate the crude adduct. This crude material is then subjected to the acidic reflux conditions without further purification, demonstrating the robustness of the intermediate, before a final workup involving neutralization and Boc-protection yields the target compound. Detailed standardized operating procedures for scaling this reaction from laboratory to pilot plant are essential to maintain consistency.
- Perform nucleophilic addition of propionitrile to keto ester using K2CO3 in acetonitrile at 0°C to room temperature.
- Execute ring-closing reaction by refluxing the adduct in concentrated hydrochloric acid overnight.
- Purify the crude product using di-tert-butyl dicarbonate and standard extraction techniques to obtain the final derivative.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain leaders, the adoption of this novel synthetic route translates directly into enhanced supply security and significant cost optimization opportunities across the value chain. By eliminating the reliance on eleven distinct synthetic steps and replacing them with a concise three-stage process, the manufacturing timeline is drastically compressed, allowing for faster turnover of production assets and reduced inventory holding costs. The substitution of sensitive, hard-to-source propiolamide with commodity-grade propionitrile removes a critical single point of failure in the supply chain, ensuring that raw material availability is no longer a bottleneck for production scheduling. Furthermore, the use of common solvents like acetonitrile and dichloromethane, along with standard inorganic bases, means that the process can be executed in existing multipurpose reactors without the need for specialized equipment or extensive retrofitting investments.
- Cost Reduction in Manufacturing: The most immediate financial benefit arises from the drastic reduction in process mass intensity, as fewer steps mean significantly lower consumption of solvents, reagents, and energy utilities per kilogram of finished product. Eliminating the need for cryogenic cooling and sensitive reagent handling also reduces the operational expenditure associated with specialized safety protocols and waste disposal fees. The higher overall yield achieved through this streamlined route means that less starting material is required to produce the same amount of active intermediate, directly lowering the variable cost of goods. Additionally, the simplified purification strategy reduces the burden on downstream processing teams, allowing for more efficient use of chromatography columns and crystallization tanks.
- Enhanced Supply Chain Reliability: The robustness of the new chemistry ensures that production campaigns are less susceptible to delays caused by reagent instability or failed batches, providing a more predictable delivery schedule for downstream customers. Since the raw materials are bulk chemicals with established global supply networks, the risk of supply disruption due to vendor-specific issues is minimized, enhancing the resilience of the procurement strategy. The ability to run the reaction at near-ambient temperatures for the initial step and standard reflux for the second step allows for greater flexibility in plant scheduling, as the process does not compete for limited cryogenic or high-pressure resources. This reliability is crucial for maintaining continuous supply lines for critical pharmaceutical programs.
- Scalability and Environmental Compliance: The process is inherently designed for scale-up, utilizing reaction conditions that are easily replicated in large-scale stainless steel reactors commonly found in CDMO facilities. The reduction in the number of isolation steps significantly decreases the volume of hazardous waste generated, aligning with modern green chemistry principles and reducing the environmental footprint of the manufacturing site. The use of hydrochloric acid for cyclization generates salt byproducts that are easier to treat and dispose of compared to the complex organic waste streams associated with multi-step transition metal catalysis. This environmental compatibility simplifies the permitting process for new production lines and supports corporate sustainability goals without compromising on output volume.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented synthesis technology in a GMP environment. These answers are derived directly from the experimental data and beneficial effects reported in the patent documentation, providing a clear picture of the technology's readiness for industrial adoption. Understanding these details is vital for technical teams evaluating the feasibility of integrating this route into their existing portfolio of intermediates.
Q: What are the primary advantages of the new synthesis route over conventional methods?
A: The new route significantly shortens the synthetic pathway compared to the traditional 11-step method, utilizes safer and more accessible raw materials like propionitrile instead of sensitive propiolamide, and offers better reaction controllability with higher overall yields.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the process is designed for scalability. It employs standard industrial solvents like acetonitrile and dichloromethane, uses common bases like potassium carbonate, and avoids cryogenic conditions or highly unstable intermediates, making it ideal for commercial scale-up.
Q: How does this method impact the purity profile of the final intermediate?
A: By reducing the number of synthetic steps and avoiding sensitive reagents that often lead to side reactions, this method minimizes the formation of complex impurity profiles, thereby facilitating easier purification and ensuring high-purity specifications required for pharmaceutical applications.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-hydroxy pyridino-azepine derivative Supplier
At NINGBO INNO PHARMCHEM, we recognize the strategic importance of efficient intermediate synthesis in accelerating the global drug development pipeline, and we are uniquely positioned to leverage this patented technology for our partners. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to industrial reactor is seamless and compliant with international quality standards. We maintain stringent purity specifications and operate rigorous QC labs equipped with state-of-the-art analytical instrumentation to guarantee that every batch of 2-hydroxy pyridino-azepine derivative meets the exacting requirements of pharmaceutical clients. Our commitment to technical excellence means we can navigate the complexities of this chemistry to deliver consistent, high-quality material that supports your clinical and commercial needs.
We invite you to engage with our technical procurement team to discuss how this optimized synthesis route can drive value for your specific projects. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into the economic benefits of switching to this streamlined process for your supply chain. We encourage you to contact us today to obtain specific COA data and route feasibility assessments tailored to your volume requirements, ensuring a partnership built on transparency, quality, and mutual success.