Optimized Synthetic Route for FTY-720 Precursor: Enhancing Yield and Scalability for Global Supply Chains
Introduction to Advanced Immunosuppressant Intermediate Synthesis
The pharmaceutical industry continuously seeks robust manufacturing pathways for complex active pharmaceutical ingredients (APIs) and their critical precursors. Patent CN1144779C introduces a groundbreaking methodology for the preparation of 2-[2-(4-alkylphenyl)-ethyl]-2-amino-propanediol, a pivotal structural motif found in potent immunosuppressive agents such as FTY-720. This technical disclosure represents a significant leap forward in process chemistry, addressing long-standing inefficiencies in the production of this high-value pharmaceutical intermediate. By shifting away from fragile, multi-step protection-deprotection strategies, the patented route leverages stable alkylbenzene derivatives to achieve a streamlined six-step synthesis. For R&D directors and procurement specialists, this innovation translates directly into enhanced process reliability and substantial economic benefits. The method not only simplifies operational complexity but also delivers a marked improvement in overall reaction yield, positioning it as a superior choice for commercial-scale manufacturing of complex amino alcohol derivatives.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art methodologies, specifically those disclosed in earlier international applications like PCT/JP93/01515, relied heavily on protected phenylethyl alcohol as the foundational starting material. This approach suffered from inherent chemical instability, particularly during the critical Friedel-Crafts acylation step involving octanoyl chloride. The instability of the phenylethyl alcohol acetate compound frequently led to side reactions and decomposition, resulting in disappointingly low yields for the acylation step. Consequently, the entire synthetic sequence, which spanned ten distinct reaction steps, culminated in a cumulative overall yield of merely approximately 5% based on the final salt form. Such inefficiency renders the conventional route economically unviable for large-scale production, creating significant supply chain vulnerabilities and inflating the cost of goods sold (COGS) for the final immunosuppressant drug. The reliance on unstable intermediates also complicates quality control, increasing the risk of batch-to-batch variability.
The Novel Approach
In stark contrast, the novel approach detailed in CN1144779C fundamentally reengineers the synthetic logic by initiating the sequence with robust alkylbenzene derivatives. This strategic shift eliminates the need for unstable protected alcohol starting materials, thereby stabilizing the early stages of the synthesis. The new route condenses the process into just six highly efficient steps, utilizing well-established reactions such as Friedel-Crafts acylation, alpha-halogenation, and malonate condensation. By optimizing reaction conditions and selecting superior reagents, this methodology achieves a total reaction yield of approximately 20-25% based on the final salt form—a four-fold improvement over the prior art. This dramatic increase in efficiency not only reduces waste and solvent consumption but also significantly shortens the production cycle time. For supply chain managers, this means a more predictable and reliable source of high-purity intermediates, capable of meeting the rigorous demands of global pharmaceutical markets without the bottlenecks associated with legacy synthesis routes.
Mechanistic Insights into the Optimized Six-Step Cascade
The core of this technological advancement lies in the precise execution of a cascade of organic transformations that maximize atom economy and minimize purification burdens. The process initiates with a classic Friedel-Crafts acylation where an alkylbenzene reacts with acetyl chloride in the presence of a Lewis acid catalyst, typically aluminum chloride (AlCl3), to generate p-alkylacetophenone with high regioselectivity. This ketone intermediate is subsequently subjected to alpha-halogenation, preferably using bromine, to introduce a reactive leaving group at the alpha position, forming the crucial p-alkyl alpha-haloacetophenone. The third and perhaps most critical step involves the condensation of this haloketone with diethyl acetamidomalonate under alkaline conditions. This step constructs the quaternary carbon center essential for the biological activity of the final molecule. Following condensation, the keto-ester intermediate undergoes a highly chemoselective reduction, preferably using sodium borohydride (NaBH4), to establish the necessary hydroxyl stereochemistry while preserving the amide functionality. The sequence concludes with hydrolysis to remove the ester and amide protecting groups, followed by catalytic hydrogenolysis to finalize the amino alcohol structure.
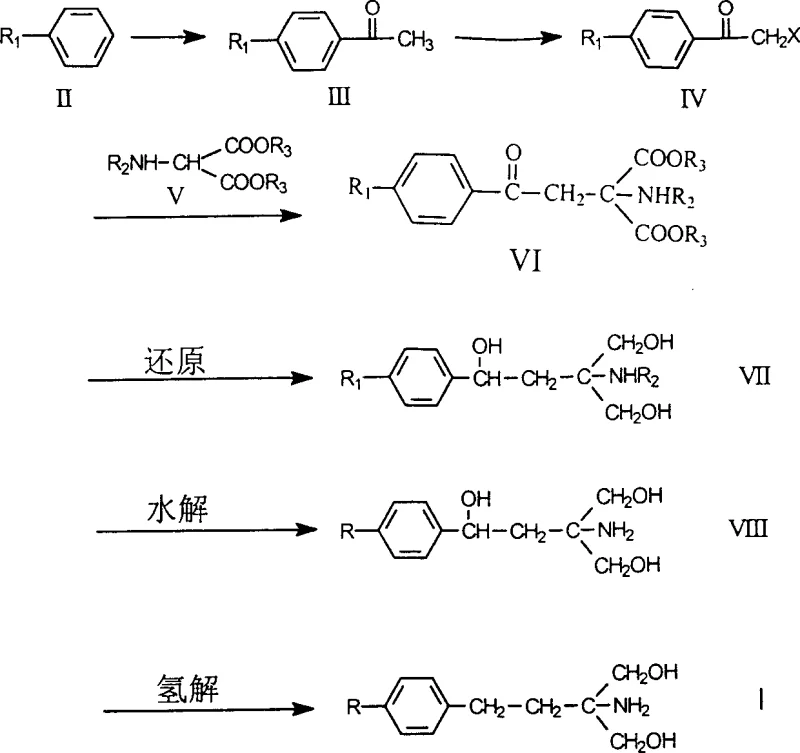
Impurity control is meticulously managed throughout this mechanistic pathway. The use of stable alkylbenzene starting materials prevents the formation of degradation products common in alcohol-based routes. Furthermore, the selection of sodium borohydride for the reduction step offers a safer and more controllable alternative to aggressive reducing agents like lithium aluminum hydride, minimizing the risk of over-reduction or side reactions that could compromise the purity profile. The final hydrogenolysis step, typically employing palladium on carbon (Pd-C) under controlled hydrogen pressure, ensures the clean removal of benzyl-type protecting groups if present, or simply finalizes the saturation state without affecting the sensitive amino-diol core. This rigorous control over reaction parameters ensures that the final product meets stringent pharmaceutical specifications, reducing the burden on downstream purification processes and ensuring consistent quality for the reliable pharmaceutical intermediate supplier network.
How to Synthesize 2-Amino-2-[2-(4-Octylphenyl)Ethyl]-1,3-Propanediol Efficiently
Implementing this synthesis requires careful attention to reaction stoichiometry and temperature control, particularly during the exothermic Friedel-Crafts and halogenation steps. The protocol dictates the use of anhydrous conditions for the initial acylation to prevent catalyst deactivation, followed by a controlled addition of halogen to manage heat evolution. The condensation step utilizes alkoxide bases such as sodium ethoxide, requiring precise molar ratios to drive the reaction to completion while minimizing dialkylation byproducts. Subsequent reduction and hydrolysis steps are performed in aqueous-alcoholic media, facilitating easy workup and isolation. The detailed standardized operating procedures for scaling this route from laboratory to pilot plant are critical for maintaining the high yields reported in the patent examples. For technical teams looking to adopt this methodology, the following guide outlines the critical operational parameters.
- Perform Friedel-Crafts acylation of alkylbenzene with acetyl chloride using AlCl3 catalyst to form p-alkylacetophenone.
- Conduct alpha-halogenation of the ketone intermediate using bromine and Lewis acid to generate alpha-haloacetophenone.
- Condense the halo-ketone with diethyl acetamidomalonate under basic conditions, followed by reduction with NaBH4, hydrolysis, and final catalytic hydrogenolysis.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this optimized synthetic route offers profound advantages that extend far beyond simple chemical yield improvements. For procurement managers tasked with cost reduction in pharmaceutical intermediates manufacturing, the elimination of four synthetic steps represents a direct reduction in labor, energy, and solvent costs. The shift to cheaper, more abundant starting materials like alkylbenzene and acetyl chloride further drives down the raw material cost base, making the final API more competitive in the global marketplace. Additionally, the simplified workflow reduces the requirement for specialized equipment and complex isolation procedures, lowering the capital expenditure barrier for contract manufacturing organizations (CMOs). This economic efficiency allows for more flexible pricing strategies and improved margins for downstream drug developers, ensuring the long-term viability of the therapeutic program.
- Cost Reduction in Manufacturing: The transition from a ten-step to a six-step process inherently reduces the cumulative loss of material at each stage, significantly boosting the mass efficiency of the entire operation. By avoiding the use of expensive and unstable protected phenylethyl alcohol precursors, the process eliminates the need for costly stabilization measures and low-yield acylation steps. The use of commodity chemicals such as aluminum chloride and sodium borohydride ensures that reagent costs remain low and predictable. Furthermore, the higher overall yield means less waste generation, which translates to reduced costs associated with waste disposal and environmental compliance. These factors combine to create a substantially leaner manufacturing cost structure.
- Enhanced Supply Chain Reliability: Supply chain resilience is critically dependent on the availability and stability of raw materials. This new route relies on alkylbenzenes, which are widely available petrochemical derivatives with stable supply chains, unlike the niche protected alcohols required by older methods. The robustness of the intermediates, particularly the acetophenone derivatives, allows for longer storage times and easier transportation without degradation risks. This stability reduces the likelihood of production delays caused by raw material spoilage or quality failures. Consequently, manufacturers can maintain tighter inventory controls and offer more reliable lead times to their clients, ensuring uninterrupted production of the final immunosuppressant medication.
- Scalability and Environmental Compliance: The simplicity of the reaction conditions facilitates seamless scale-up from kilogram to multi-ton production scales. The avoidance of hazardous reagents where possible, and the use of standard solvents like ethanol and ethyl acetate, aligns well with modern green chemistry principles. The reduction in step count inherently lowers the E-factor (mass of waste per mass of product), contributing to a smaller environmental footprint. This alignment with sustainability goals is increasingly important for regulatory approval and corporate social responsibility mandates. The process is designed to be easily adapted to continuous flow chemistry or large batch reactors, ensuring that commercial scale-up of complex pharmaceutical intermediates can be achieved with minimal technical risk.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented synthesis route. These insights are derived directly from the experimental data and comparative analysis provided in the patent documentation, offering clarity on yield expectations, reagent selection, and process safety. Understanding these nuances is essential for technical teams evaluating the feasibility of technology transfer. The answers below reflect the consensus of the patent's teachings regarding optimal conditions and potential variations.
Q: What is the primary advantage of this synthesis route over prior art methods?
A: The primary advantage is the significant reduction in synthetic steps from 10 to 6, which eliminates the use of unstable protected phenylethyl alcohol starting materials. This modification increases the overall yield from approximately 5% to 20-25%, drastically improving process economics and scalability.
Q: Which reducing agents are preferred for the conversion of intermediate VI to VII?
A: While various complex hydrides like LiAlH4 can be used, sodium borohydride (NaBH4) is the most preferred reducing agent due to its operational safety, cost-effectiveness, and high chemoselectivity in reducing the ketone functionality without affecting other sensitive groups in the molecule.
Q: How does this process impact supply chain stability for immunosuppressant production?
A: By utilizing stable, commercially available starting materials like alkylbenzene and acetyl chloride, the process removes bottlenecks associated with synthesizing unstable precursors. This ensures consistent raw material availability and reduces the risk of batch failures, leading to more reliable delivery schedules for downstream API manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Amino-2-[2-(4-Octylphenyl)Ethyl]-1,3-Propanediol Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient synthesis routes in the development of life-saving immunosuppressants. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of patent CN1144779C are fully realized in practical manufacturing environments. We adhere to stringent purity specifications and operate rigorous QC labs to guarantee that every batch of intermediate meets the highest global standards. Our commitment to process optimization allows us to deliver high-purity intermediates that facilitate smoother downstream API synthesis, reducing the burden on our partners' formulation teams.
We invite pharmaceutical companies and research institutions to collaborate with us to leverage this advanced technology for their supply chains. By partnering with us, you gain access to a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments. Together, we can accelerate the availability of affordable, high-quality immunosuppressive therapies for patients worldwide, leveraging our expertise as a trusted partner in fine chemical manufacturing.