Scalable Synthesis of Novel N-Acetylciprofloxacin Propenone Derivatives for Oncology Applications
The pharmaceutical landscape is constantly evolving towards hybrid molecules that combine distinct pharmacophores to overcome resistance and improve therapeutic indices. Patent CN111646937A introduces a groundbreaking class of N-acetylciprofloxacin propenone derivatives, effectively merging the robust fluoroquinolone skeleton with the potent antitumor properties of the chalcone motif. This strategic molecular design addresses critical limitations found in existing therapies, particularly regarding drug resistance and solubility profiles. By leveraging the well-established topoisomerase inhibition mechanism of fluoroquinolones and integrating it with the apoptosis-inducing capabilities of arylpropenone structures, this technology offers a compelling avenue for next-generation oncology drug development. The resulting compounds exhibit a unique structural architecture that not only enhances cytotoxicity against various tumor lines but also demonstrates reduced toxicity towards normal cells, a paramount consideration in clinical translation.
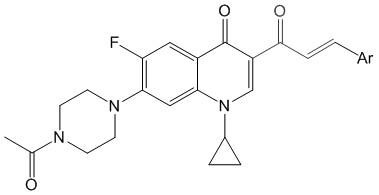
For procurement specialists and supply chain managers, the accessibility of the starting materials presents a significant logistical advantage. The synthesis begins with N-acetylciprofloxacin, a commercially available and cost-effective bulk pharmaceutical chemical, ensuring a stable and reliable supply chain foundation. Unlike complex natural product extractions which are subject to agricultural variability and seasonal fluctuations, this semi-synthetic approach relies on consistent industrial fermentation products. This stability translates directly into predictable lead times and reduced risk of raw material shortages, making it an ideal candidate for reliable pharmaceutical intermediate supplier partnerships aiming for long-term production continuity.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional approaches to developing antitumor agents often rely heavily on natural chalcones or simple fluoroquinolone derivatives, both of which possess inherent drawbacks that limit their clinical utility. Natural chalcones, while biologically active, are notoriously plagued by poor aqueous solubility, leading to low bioavailability and erratic absorption rates in vivo. This physicochemical limitation necessitates complex formulation strategies or high dosing regimens, which can exacerbate off-target toxicity and increase manufacturing costs. Furthermore, simple fluoroquinolone modifications at the C-3 position have historically yielded mixed results, with many derivatives failing to demonstrate a significant improvement in antitumor potency over their antibacterial progenitors. The lack of a dual-mechanism approach in conventional single-scaffold drugs often allows tumor cells to develop resistance mechanisms more rapidly, rendering the therapy ineffective over time.
The Novel Approach
The methodology outlined in CN111646937A circumvents these issues through a rational pharmacophore splicing strategy. By covalently linking the fluoroquinolone core to an arylpropenone moiety, the invention creates a 'chalcone-like' hybrid that inherits the best properties of both parents. The introduction of the hydrophilic piperazine ring from the ciprofloxacin backbone dramatically improves the water solubility of the chalcone fragment, thereby enhancing bioavailability without the need for exotic solubilizing excipients. Moreover, this hybridization allows for simultaneous targeting of topoisomerase enzymes and other cellular pathways affected by chalcones, creating a multi-target therapeutic effect that complicates the development of drug resistance. This structural innovation represents a significant leap forward in cost reduction in pharmaceutical intermediate manufacturing by maximizing the therapeutic value of each synthesized molecule.
Mechanistic Insights into Claisen-Schmidt Condensation and Pharmacophore Splicing
The core chemical transformation enabling this technology is a base-catalyzed Claisen-Schmidt condensation, a robust and atom-economical reaction widely favored in industrial organic synthesis. In the final step of the sequence, the C-3 acetyl ketone intermediate reacts with various aromatic aldehydes to form the conjugated enone system characteristic of the propenone derivatives. This reaction proceeds under mild conditions using common organic bases such as piperidine or triethylamine in ethanol, avoiding the need for harsh reagents or extreme temperatures that could degrade the sensitive fluoroquinolone core. The mechanistic pathway involves the formation of an enolate ion from the ketone, which then nucleophilically attacks the carbonyl carbon of the aldehyde, followed by dehydration to establish the thermodynamically stable trans-double bond configuration. This high degree of stereocontrol is crucial for maintaining the biological activity of the final product.
Beyond the final coupling, the preceding steps involve precise activation and functionalization of the quinolone ring. The initial conversion of the C-3 carboxylic acid to an imidazole amide using carbonyldiimidazole (CDI) serves as a critical activation step, transforming a relatively inert acid into a highly reactive acylating agent. This activated intermediate then undergoes condensation with potassium monoethyl malonate in the presence of magnesium chloride, a Lewis acid that facilitates the enolization and subsequent attack on the amide carbonyl. The subsequent hydrolysis and decarboxylation steps are meticulously controlled to ensure the selective formation of the methyl ketone without affecting the cyclopropyl group at N-1 or the piperazine substituent at C-7. This rigorous control over the impurity profile is essential for meeting the stringent purity specifications required for clinical-grade materials, ensuring that the final API intermediates are free from genotoxic impurities or residual starting materials.
How to Synthesize N-Acetylciprofloxacin Propenone Derivatives Efficiently
The synthesis of these high-value antitumor candidates follows a streamlined four-step linear sequence that is highly amenable to scale-up. The process begins with the activation of the starting material, followed by chain extension, decarboxylation, and finally the condensation with the desired aldehyde. Each step has been optimized to maximize yield and minimize purification complexity, utilizing standard unit operations such as reflux, filtration, and recrystallization. The detailed standardized synthesis steps, including specific molar ratios, solvent choices, and reaction times, are provided in the guide below to assist process chemists in replicating this high-efficiency route.
- Activate N-acetylciprofloxacin with carbonyldiimidazole (CDI) in anhydrous acetonitrile to form the imidazole amide intermediate.
- Perform condensation with potassium monoethyl malonate using MgCl2 and triethylamine to introduce the beta-keto ester side chain.
- Hydrolyze and decarboxylate the ester intermediate using aqueous sodium hydroxide to yield the C-3 acetyl ketone precursor.
- Execute Claisen-Schmidt condensation with various aromatic aldehydes under basic catalysis to finalize the propenone derivative structure.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, this synthetic route offers substantial benefits that align perfectly with the goals of modern pharmaceutical supply chains focused on resilience and cost efficiency. The reliance on commodity chemicals and avoidance of precious metal catalysts significantly lowers the direct material costs associated with production. Furthermore, the operational simplicity of the reactions reduces the burden on manufacturing infrastructure, allowing for production in standard glass-lined or stainless steel reactors without the need for specialized high-pressure or cryogenic equipment. This accessibility lowers the barrier to entry for contract manufacturing organizations and ensures that the supply of these critical intermediates can be rapidly scaled to meet market demand without prohibitive capital expenditure.
- Cost Reduction in Manufacturing: The elimination of transition metal catalysts such as palladium or platinum from the synthetic route removes the necessity for expensive metal scavenging processes and rigorous heavy metal testing, which are significant cost drivers in API production. Additionally, the use of ethanol and acetonitrile as primary solvents allows for efficient recovery and recycling systems, further driving down the variable costs per kilogram. The high yields reported in the patent examples suggest a material-efficient process that minimizes waste generation, contributing to a leaner and more economically viable manufacturing model that supports competitive pricing strategies for the final drug product.
- Enhanced Supply Chain Reliability: The starting material, N-acetylciprofloxacin, is produced on a multi-ton scale globally for veterinary and human use, ensuring a robust and diversified supply base that mitigates the risk of single-source dependency. The aromatic aldehydes required for the final condensation step are also ubiquitous fine chemicals available from numerous suppliers worldwide, providing procurement teams with ample flexibility to negotiate favorable terms and secure backup sources. This redundancy in the supply chain is critical for maintaining continuous production schedules and preventing disruptions that could delay clinical trials or commercial launches, thereby safeguarding the project timeline against external market volatility.
- Scalability and Environmental Compliance: The synthetic pathway utilizes reagents and conditions that are well-understood in the context of Good Manufacturing Practice (GMP) production, facilitating a smoother technology transfer from laboratory to pilot and commercial scales. The absence of hazardous reagents like thionyl chloride or strong mineral acids in the key coupling steps simplifies waste treatment protocols and reduces the environmental footprint of the manufacturing process. This alignment with green chemistry principles not only aids in regulatory compliance but also enhances the corporate sustainability profile of the manufacturing partner, a factor that is increasingly weighted in vendor selection criteria by major pharmaceutical companies.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these novel derivatives. The answers are derived directly from the experimental data and technical specifications disclosed in the patent literature, providing a factual basis for decision-making. Understanding these details is crucial for R&D teams evaluating the feasibility of incorporating this scaffold into their drug discovery pipelines and for supply chain professionals assessing the logistical requirements for sourcing these materials.
Q: What is the primary advantage of these propenone derivatives over natural chalcones?
A: Unlike natural chalcones which suffer from poor water solubility and low bioavailability, these N-acetylciprofloxacin hybrids incorporate a hydrophilic piperazine group, significantly enhancing solubility and biological uptake while maintaining potent antitumor activity.
Q: Does this synthesis route require expensive transition metal catalysts?
A: No, the key coupling steps utilize magnesium chloride and organic bases like piperidine or triethylamine. This avoids the need for costly palladium or other transition metal catalysts, simplifying purification and reducing heavy metal contamination risks.
Q: What specific cancer cell lines have shown sensitivity to these compounds?
A: The compounds demonstrate significant inhibitory activity against human non-small cell lung cancer (A549), renal cancer (769-P), liver cancer (Hep-3B), and notably show efficacy against sunitinib-resistant renal cell carcinoma lines, indicating potential for overcoming drug resistance.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable N-Acetylciprofloxacin Derivatives Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of the hybrid molecular architecture described in CN111646937A for the future of oncology therapeutics. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from preclinical research to clinical supply is seamless and efficient. Our state-of-the-art facilities are equipped to handle the specific unit operations required for this synthesis, including precise temperature control for reflux reactions and advanced crystallization technologies to achieve stringent purity specifications. With our rigorous QC labs and commitment to quality, we guarantee that every batch of N-acetylciprofloxacin propenone derivatives meets the highest international standards for safety and efficacy.
We invite you to collaborate with us to optimize this synthesis for your specific needs, whether you require custom analogs with different aromatic substituents or large-scale production of the lead compounds. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your project volume, helping you identify opportunities for further process intensification and cost optimization. Please contact us today to request specific COA data and route feasibility assessments, and let us demonstrate how our expertise can accelerate your drug development timeline while controlling costs.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →