Scalable Synthesis of 1-Cyclopropyl Quinolone Carboxylic Acid Derivatives for Pharmaceutical Applications
The pharmaceutical industry continuously seeks robust and scalable pathways for the production of high-value antibiotic intermediates, particularly those belonging to the fluoroquinolone class. Patent CN1037147A discloses a sophisticated methodology for the preparation of 1-cyclopropyl quinolone carboxylic acid derivatives, which serve as critical precursors for potent antimicrobial agents like ciprofloxacin. The core innovation lies in the strategic construction of the quinolone nucleus through a novel N-cyclopropyl aniline intermediate, offering a distinct advantage over traditional alkylation methods that often suffer from poor regioselectivity or harsh reaction conditions. This technical disclosure provides a comprehensive framework for synthesizing compounds of Formula (I), where the 1-position is substituted with a cyclopropyl group, a structural motif known to significantly enhance biological activity and metabolic stability. 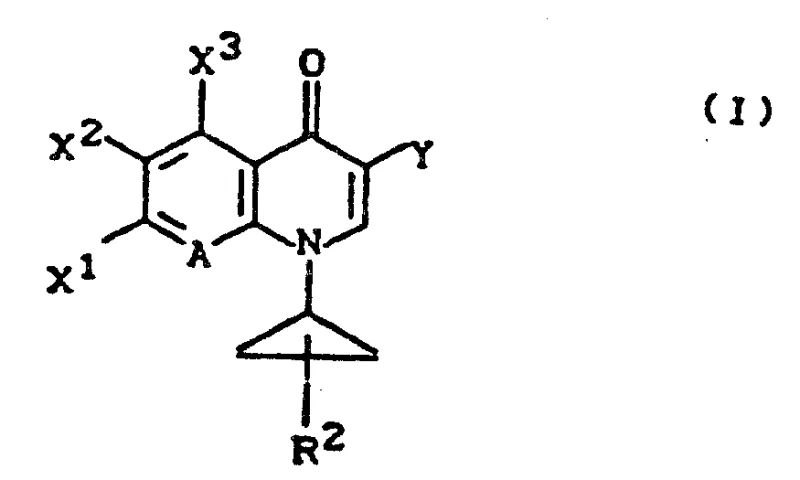 For R&D directors and process chemists, understanding the versatility of this route is paramount, as it accommodates various substituents at the 6, 7, and 8 positions, allowing for the generation of diverse libraries of potential drug candidates. The patent emphasizes that the intermediate N-cyclopropyl aniline (IV) is itself a novel compound, representing a valuable building block that can be isolated and characterized before proceeding to the final cyclization steps. By leveraging this specific synthetic architecture, manufacturers can achieve higher purity profiles and more consistent batch-to-batch reproducibility, which are essential metrics for regulatory compliance in API manufacturing.
For R&D directors and process chemists, understanding the versatility of this route is paramount, as it accommodates various substituents at the 6, 7, and 8 positions, allowing for the generation of diverse libraries of potential drug candidates. The patent emphasizes that the intermediate N-cyclopropyl aniline (IV) is itself a novel compound, representing a valuable building block that can be isolated and characterized before proceeding to the final cyclization steps. By leveraging this specific synthetic architecture, manufacturers can achieve higher purity profiles and more consistent batch-to-batch reproducibility, which are essential metrics for regulatory compliance in API manufacturing.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the introduction of a cyclopropyl group onto the nitrogen atom of a quinolone or aniline precursor has presented significant synthetic challenges that impact both cost and efficiency. Conventional alkylation strategies often rely on the direct reaction of aniline with cyclopropyl halides, which frequently requires elevated temperatures and strong bases that can lead to undesirable side reactions, such as ring-opening of the cyclopropane moiety or poly-alkylation of the amine. These side reactions not only reduce the overall yield of the desired mono-alkylated product but also generate complex impurity profiles that are difficult and expensive to remove during downstream purification. Furthermore, traditional routes may involve multiple protection and de-protection steps to ensure selectivity, adding unnecessary unit operations that increase solvent consumption, waste generation, and total processing time. For procurement managers, these inefficiencies translate directly into higher raw material costs and longer lead times, creating bottlenecks in the supply chain for critical antibiotic intermediates. The reliance on harsh conditions also poses safety risks and limits the choice of compatible functional groups, restricting the chemical space that can be explored for new derivative development.
The Novel Approach
In contrast, the methodology outlined in CN1037147A introduces a streamlined and chemoselective approach that circumvents these traditional pitfalls by utilizing specific cyclopropane derivatives of Formula (III). This novel route employs 1-halo-1-alkoxy or 1-halo-1-thioalkyl cyclopropanes, which act as superior electrophiles for the nucleophilic attack by the aniline nitrogen. The presence of the alkoxy or thioalkyl leaving group facilitates the substitution reaction under much milder thermal conditions, typically ranging from 40°C to 80°C, thereby preserving the integrity of the sensitive cyclopropane ring. This gentle reaction environment minimizes the formation of byproducts and eliminates the need for aggressive reagents, resulting in a cleaner reaction profile and simplified workup procedures. For supply chain heads, this translates to a more reliable manufacturing process with reduced risk of batch failures due to thermal runaway or impurity accumulation. The ability to perform this key N-cyclopropylation step efficiently sets the stage for a high-yielding subsequent cyclization, ensuring that the overall process economics are favorable for commercial scale-up of complex pharmaceutical intermediates without compromising on quality or safety standards.
Mechanistic Insights into N-Cyclopropylation and Cyclization
The mechanistic pathway begins with the nucleophilic substitution reaction between a substituted aniline of Formula (II) and a cyclopropane derivative of Formula (III), leading to the formation of the key N-cyclopropyl aniline intermediate (IV). 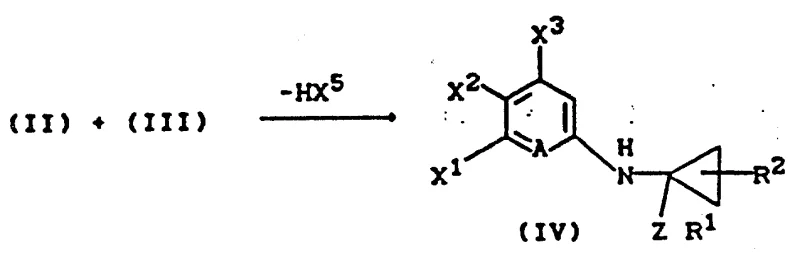 In this step, the lone pair of electrons on the aniline nitrogen attacks the electrophilic carbon of the cyclopropane ring, displacing the halogen atom (X5) and the ZR1 group, often facilitated by an acid scavenger such as triethylamine or pyridine. The choice of solvent is critical here; non-polar or moderately polar solvents like dichloromethane, toluene, or chloroform are preferred to solubilize the reactants while maintaining the stability of the transition state. Following the formation of intermediate (IV), a reduction step is employed to convert the protected amine into the free N-cyclopropyl aniline (V), typically using sodium borohydride in the presence of boron trifluoride etherate. This reduction is highly specific, cleaving the ether or thioether linkage without affecting other sensitive functional groups on the aromatic ring, such as nitro or halogen substituents, which is crucial for maintaining the structural integrity required for the final antibiotic activity. The precision of this reduction step ensures that the resulting amine is ready for the final condensation without requiring extensive purification, thereby enhancing the overall atom economy of the process.
In this step, the lone pair of electrons on the aniline nitrogen attacks the electrophilic carbon of the cyclopropane ring, displacing the halogen atom (X5) and the ZR1 group, often facilitated by an acid scavenger such as triethylamine or pyridine. The choice of solvent is critical here; non-polar or moderately polar solvents like dichloromethane, toluene, or chloroform are preferred to solubilize the reactants while maintaining the stability of the transition state. Following the formation of intermediate (IV), a reduction step is employed to convert the protected amine into the free N-cyclopropyl aniline (V), typically using sodium borohydride in the presence of boron trifluoride etherate. This reduction is highly specific, cleaving the ether or thioether linkage without affecting other sensitive functional groups on the aromatic ring, such as nitro or halogen substituents, which is crucial for maintaining the structural integrity required for the final antibiotic activity. The precision of this reduction step ensures that the resulting amine is ready for the final condensation without requiring extensive purification, thereby enhancing the overall atom economy of the process.
The final stage involves the condensation of the reduced amine (V) with an ene derivative of Formula (VI), such as ethoxy methylene diethyl malonate, followed by a thermal cyclization to close the quinolone ring. 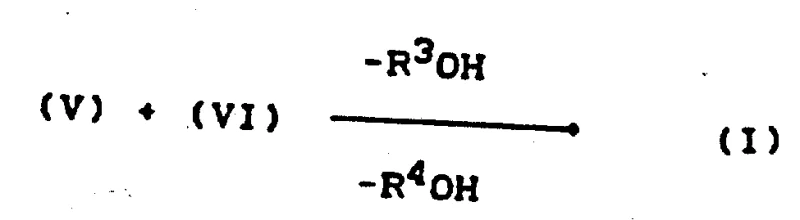 This condensation reaction forms an enamine intermediate, which subsequently undergoes an intramolecular cyclization upon heating to high temperatures (100-250°C), often in high-boiling solvents like diphenyl ether or biphenyl. During this cyclization, the ester or alkoxy groups (R3OH and R4OH) are eliminated, driving the formation of the rigid 4-quinolone-3-carboxylic acid skeleton. The mechanism relies on the precise alignment of the nucleophilic nitrogen and the electrophilic beta-carbon of the ene derivative, a process that is highly favored by the electron-withdrawing nature of the ester groups. For R&D teams, understanding this mechanism allows for the optimization of reaction parameters, such as temperature ramping and solvent selection, to maximize the conversion to the desired regioisomer while minimizing the formation of decarboxylated byproducts. The robustness of this cyclization step ensures that even with varying substituents on the aniline ring, the core quinolone structure is formed reliably, providing a versatile platform for the synthesis of a wide range of fluoroquinolone antibiotics.
This condensation reaction forms an enamine intermediate, which subsequently undergoes an intramolecular cyclization upon heating to high temperatures (100-250°C), often in high-boiling solvents like diphenyl ether or biphenyl. During this cyclization, the ester or alkoxy groups (R3OH and R4OH) are eliminated, driving the formation of the rigid 4-quinolone-3-carboxylic acid skeleton. The mechanism relies on the precise alignment of the nucleophilic nitrogen and the electrophilic beta-carbon of the ene derivative, a process that is highly favored by the electron-withdrawing nature of the ester groups. For R&D teams, understanding this mechanism allows for the optimization of reaction parameters, such as temperature ramping and solvent selection, to maximize the conversion to the desired regioisomer while minimizing the formation of decarboxylated byproducts. The robustness of this cyclization step ensures that even with varying substituents on the aniline ring, the core quinolone structure is formed reliably, providing a versatile platform for the synthesis of a wide range of fluoroquinolone antibiotics.
How to Synthesize 1-Cyclopropyl Quinolone Derivatives Efficiently
The synthesis of these high-value intermediates requires precise control over reaction stoichiometry and thermal conditions to ensure optimal yield and purity. The process generally follows a three-stage sequence: initial N-cyclopropylation, reduction of the intermediate, and final condensation-cyclization. Detailed operational parameters, including specific molar ratios, solvent volumes, and temperature profiles for each stage, are critical for successful replication and scale-up.
- React substituted aniline with a cyclopropane derivative (e.g., 1-bromo-1-ethoxycyclopropane) in the presence of an acid scavenger to form N-cyclopropyl aniline.
- Reduce the N-cyclopropyl aniline intermediate using a reducing agent like sodium borohydride mixed with boron trifluoride etherate to remove protecting groups.
- Condense the reduced amine with an ene derivative (e.g., ethoxy methylene diethyl malonate) followed by thermal cyclization to yield the final quinolone structure.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this synthetic route offers substantial benefits for procurement managers and supply chain leaders focused on cost efficiency and reliability. The primary advantage lies in the simplification of the synthetic sequence, which reduces the total number of unit operations required to reach the final API intermediate. By eliminating the need for complex protection-deprotection sequences and harsh alkylation conditions, the process significantly lowers the consumption of expensive reagents and specialized catalysts. This streamlining directly contributes to cost reduction in API manufacturing, as fewer raw materials are consumed, and less energy is expended on heating and cooling cycles. Furthermore, the use of common, commercially available solvents such as dichloromethane, tetrahydrofuran, and diphenyl ether ensures that the supply chain is not dependent on niche or volatile chemical markets, enhancing supply continuity. The mild conditions of the initial N-cyclopropylation step also reduce the wear and tear on reactor equipment, extending asset life and lowering maintenance costs over time.
- Cost Reduction in Manufacturing: The elimination of transition metal catalysts and the use of inexpensive reducing agents like sodium borohydride drastically simplify the downstream purification process. Without the need for expensive heavy metal scavenging resins or complex chromatography steps to remove metal residues, the overall production cost is significantly optimized. This lean manufacturing approach allows for competitive pricing strategies while maintaining healthy margins, making the final quinolone intermediates more attractive to generic drug manufacturers. Additionally, the high selectivity of the reaction minimizes the generation of waste streams, reducing the costs associated with environmental compliance and waste disposal.
- Enhanced Supply Chain Reliability: The starting materials, including various substituted anilines and cyclopropane derivatives, are readily available from multiple global suppliers, mitigating the risk of single-source dependency. This diversity in the supply base ensures that production schedules can be maintained even if one vendor faces disruptions, providing a buffer against market volatility. The robustness of the reaction conditions means that the process is less sensitive to minor fluctuations in raw material quality, further stabilizing the supply chain. For supply chain heads, this reliability translates to predictable lead times and the ability to commit to long-term delivery contracts with confidence, securing the availability of critical antibiotic ingredients.
- Scalability and Environmental Compliance: The process is inherently scalable, having been demonstrated to work effectively from gram-scale laboratory experiments to multi-kilogram pilot runs without significant loss in efficiency. The thermal cyclization step, while requiring high temperatures, can be managed in standard stainless steel reactors equipped with appropriate heating systems, avoiding the need for specialized high-pressure equipment. From an environmental standpoint, the reduction in solvent usage and the avoidance of toxic heavy metals align with modern green chemistry principles, facilitating easier regulatory approval and reducing the carbon footprint of the manufacturing site. This alignment with sustainability goals is increasingly important for pharmaceutical companies aiming to meet corporate social responsibility targets.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis of 1-cyclopropyl quinolone derivatives based on the patented technology. These insights are derived directly from the experimental data and claims presented in the patent documentation, providing clarity on the feasibility and advantages of the method. Understanding these details helps stakeholders make informed decisions regarding process adoption and partnership opportunities.
Q: What is the primary advantage of the N-cyclopropylation method described in CN1037147A?
A: The method allows for the direct introduction of the cyclopropyl group onto the aniline nitrogen under relatively mild conditions (40-80°C), avoiding the harsh alkylation conditions often required in conventional routes, which improves yield and purity.
Q: Which reducing agents are preferred for the conversion of intermediate (IV) to (V)?
A: The patent specifies sodium cyanoborohydride or sodium borohydride, with a particular preference for a 1:1 mixture of sodium borohydride and boron trifluoride-ether complex for optimal efficiency in tetrahydrofuran.
Q: Is this synthesis route suitable for large-scale commercial production?
A: Yes, the process utilizes common industrial solvents such as dichloromethane, toluene, and diphenyl ether, and operates at manageable temperatures and pressures, making it highly adaptable for commercial scale-up of complex pharmaceutical intermediates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 1-Cyclopropyl Quinolone Supplier
The technical potential of the synthesis route described in CN1037147A is immense, offering a pathway to high-purity quinolone intermediates that are essential for the global antibiotic market. NINGBO INNO PHARMCHEM stands ready to leverage this technology, bringing extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our facility is equipped with rigorous QC labs and stringent purity specifications to ensure that every batch of 1-cyclopropyl quinolone carboxylic acid meets the highest international standards. We understand the critical nature of these intermediates in the fight against bacterial infections and are committed to delivering consistent quality that supports your regulatory filings and commercial success.
We invite you to engage with our technical procurement team to discuss how we can tailor this synthesis to your specific volume requirements. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into how our optimized process can reduce your overall COGS. We encourage potential partners to contact us for specific COA data and route feasibility assessments, ensuring that our capabilities align perfectly with your project timelines and quality expectations for reliable pharmaceutical intermediate supply.