Advanced Synthesis of Isoberaline Analogues: A Strategic Route for Anti-Tuberculosis API Development
The global fight against tuberculosis (TB) faces a critical bottleneck due to the rising prevalence of multidrug-resistant strains and the scarcity of novel therapeutic scaffolds. Addressing this urgent medical challenge requires innovative chemical strategies that merge proven pharmacophores with new structural motifs. Patent CN114276369B presents a groundbreaking methodology for the preparation of isoberaline analogues, effectively bridging the gap between fluoroquinolone antibiotics and natural indoloquinoline alkaloids. This technology leverages the commercially abundant starting material, rufloxacin, to construct a complex indolo[3,2-c]quinoline skeleton through an atom-economical pathway. For pharmaceutical developers, this represents a significant opportunity to access a new class of anti-tuberculosis agents that possess enhanced water solubility and superior resistance profiles compared to traditional cryptolepine derivatives.
The strategic value of this patent lies in its ability to transform a second-line anti-TB drug into a lead compound for a entirely new chemical entity. By retaining the hydrophilic piperazine moiety from the parent fluoroquinolone, the synthesized analogues overcome the notorious poor bioavailability associated with natural alkaloids. Furthermore, the introduction of fluorine atoms and various substituents on the indole ring allows for fine-tuning of pharmacokinetic properties. This dual-advantage approach not only solves the sourcing difficulties of natural products but also creates a robust platform for developing next-generation therapeutics capable of combating resistant Mycobacterium tuberculosis strains.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the development of cryptolepine-based anti-tuberculosis drugs has been severely hampered by reliance on natural extraction or complex total synthesis. Natural sources of cryptolepine and its isomers, such as isocryptolepine, are limited and subject to ecological variability, making large-scale supply chains unreliable for commercial API manufacturing. Moreover, these natural alkaloids suffer from intrinsic physicochemical defects, specifically poor water solubility, which leads to low oral bioavailability and limits their clinical utility. Traditional synthetic routes often involve harsh conditions, multiple protection-deprotection steps, and the use of expensive transition metal catalysts, resulting in low overall yields and high production costs. These factors collectively create a high barrier to entry for bringing cryptolepine derivatives to the market as viable pharmaceutical products.
The Novel Approach
The methodology disclosed in the patent revolutionizes this landscape by utilizing rufloxacin, a readily available fluoroquinolone, as the primary building block. This semi-synthetic strategy bypasses the need for natural extraction entirely. The core innovation involves a reductive decarboxylation followed by a Fischer indole synthesis, which efficiently constructs the target indoloquinoline framework in just two major transformation steps. This approach capitalizes on the existing functional groups within rufloxacin, specifically the piperazine ring, thereby eliminating the need for additional solubilizing modifications later in the synthesis. The result is a streamlined process that significantly reduces waste generation and operational complexity, offering a scalable solution for producing high-purity intermediates suitable for drug development.
This synthetic efficiency is further enhanced by the specific choice of reducing agents and reaction conditions. Unlike conventional methods that might rely on sensitive hydrides requiring strict anhydrous environments, this process utilizes potassium borohydride in alcoholic solvents under reflux. The subsequent cyclization with substituted phenylhydrazines proceeds smoothly under acidic catalysis, demonstrating excellent tolerance for various functional groups such as methoxy, methyl, halogen, and sulfonamide substituents. This versatility allows for the rapid generation of a diverse library of analogues, enabling medicinal chemists to optimize structure-activity relationships (SAR) without being constrained by synthetic feasibility.
Mechanistic Insights into Reductive Decarboxylation and Fischer Indole Synthesis
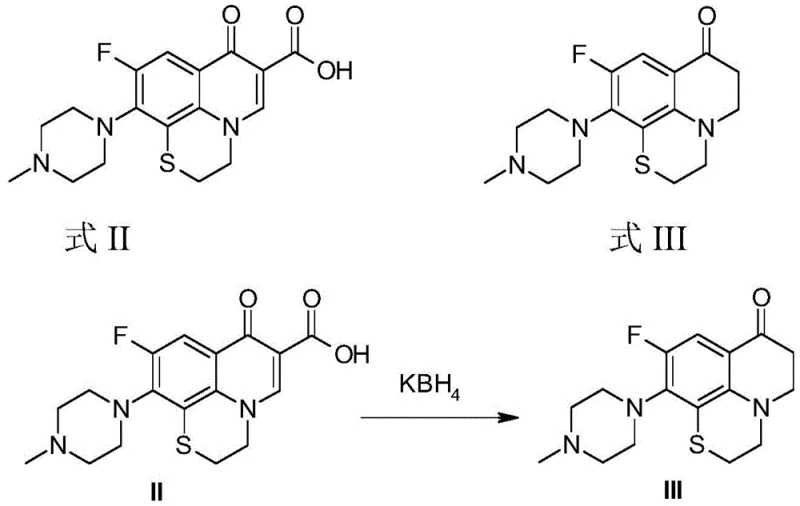
The chemical transformation begins with the reductive decarboxylation of the rufloxacin precursor (Formula II). In this critical step, the carboxylic acid group at the 3-position of the quinolone ring is removed while the 4-keto group is preserved or transiently reduced and re-oxidized depending on the workup, ultimately yielding the 2,3-dihydroquinolin-4-one intermediate (Formula III). The use of potassium borohydride (KBH4) is mechanistically significant; it acts as a source of hydride ions that attack the carbonyl or activate the decarboxylation pathway. The patent highlights that KBH4 is superior to sodium borohydride in this context due to its lower hygroscopicity and milder reactivity, which minimizes side reactions and simplifies the purification process. The reaction is typically conducted in methanol or ethanol, where the solvent also participates in proton transfer events necessary for the stabilization of the intermediate species.
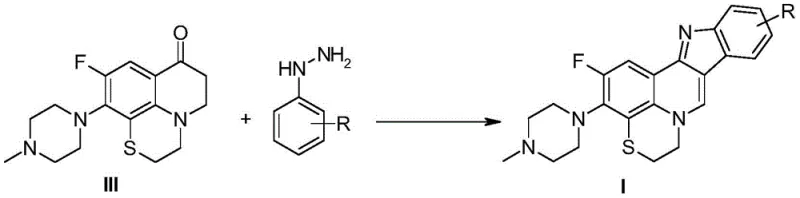
Following the formation of the quinolinone intermediate, the synthesis proceeds via a classic Fischer indole synthesis to close the second ring. The ketone functionality of the intermediate reacts with the amino group of the substituted phenylhydrazine to form a hydrazone intermediate. Under acidic catalysis (using HCl, H2SO4, or polyphosphoric acid), this hydrazone undergoes a [3,3]-sigmatropic rearrangement, followed by aromatization and elimination of ammonia to form the fused indole ring. This cascade reaction is highly efficient for constructing the rigid indolo[3,2-c]quinoline core. The mechanism ensures that the substituents on the phenylhydrazine ring (R groups) are precisely positioned on the newly formed indole moiety, allowing for targeted modulation of the molecule's electronic and steric properties. This level of control is essential for optimizing binding affinity to biological targets in Mycobacterium tuberculosis.
Impurity control is rigorously managed throughout this mechanistic pathway. The patent specifies detailed workup procedures, including pH adjustments and activated carbon treatment, to remove unreacted hydrazines and colored byproducts. Recrystallization from specific solvent systems, such as hot water followed by n-hexane for the intermediate, and ethanol-ethyl acetate mixtures for the final product, ensures that the final API intermediate meets stringent purity specifications. This focus on purification is critical for pharmaceutical applications, where trace impurities can affect safety profiles and regulatory approval.
How to Synthesize Isoberaline Analogues Efficiently
The synthesis of these high-value anti-tuberculosis intermediates follows a robust and reproducible protocol designed for scalability. The process starts with the suspension of rufloxacin in anhydrous alcohol, followed by the controlled addition of the reducing agent. After the initial reduction, the reaction mixture is worked up to isolate the quinolinone core, which then serves as the substrate for the cyclization step. The final stage involves reacting this core with the appropriate phenylhydrazine derivative under reflux conditions. Detailed standardized operating procedures for each reaction phase, including temperature controls, molar ratios, and crystallization parameters, are essential for maintaining batch-to-batch consistency. For comprehensive technical details on reaction times and specific solvent volumes, please refer to the structured guide below.
- Perform reductive decarboxylation of rufloxacin (Formula II) using potassium borohydride to obtain the 2,3-dihydroquinolin-4-one intermediate (Formula III).
- React the quinolinone intermediate with substituted phenylhydrazines via Fischer indole synthesis to construct the indoloquinoline core.
- Purify the final crude product through activated carbon decolorization and recrystallization using ethanol-ethyl acetate mixtures.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, this synthetic route offers substantial strategic benefits over traditional natural product extraction or multi-step total synthesis. The primary advantage is the reliance on rufloxacin, a mature, commercially available commodity chemical. This ensures a stable and predictable supply of raw materials, mitigating the risks associated with agricultural sourcing or complex upstream synthesis. The streamlined two-step process significantly reduces the number of unit operations required, which directly translates to lower manufacturing overheads and reduced energy consumption. By minimizing the number of isolation and purification stages, the overall process mass intensity (PMI) is improved, aligning with modern green chemistry principles and reducing waste disposal costs.
- Cost Reduction in Manufacturing: The substitution of expensive or hazardous reagents with cost-effective alternatives like potassium borohydride drives down raw material expenses. Furthermore, the high atom economy of the Fischer indole synthesis means that a larger proportion of the starting material mass is incorporated into the final product, reducing waste. The elimination of transition metal catalysts removes the need for costly and time-consuming heavy metal scavenging steps, which are often a bottleneck in API production. This simplified workflow allows for significant operational expenditure savings without compromising on the quality or purity of the final pharmaceutical intermediate.
- Enhanced Supply Chain Reliability: Utilizing a semi-synthetic route based on established fluoroquinolone chemistry ensures high supply chain resilience. Unlike natural alkaloids that are subject to seasonal and geopolitical supply fluctuations, the precursors for this synthesis are produced via established industrial processes with global capacity. The robustness of the reaction conditions, which tolerate a range of temperatures and solvent qualities, further enhances manufacturability. This reliability is crucial for long-term drug development projects where consistent material supply is mandatory for preclinical and clinical trials.
- Scalability and Environmental Compliance: The process is inherently scalable, having been demonstrated effectively from gram to multi-gram scales in the patent examples. The use of common organic solvents like ethanol and methanol simplifies solvent recovery and recycling systems, facilitating compliance with environmental regulations. The avoidance of toxic heavy metals and the generation of manageable aqueous waste streams make this route environmentally sustainable. This alignment with ESG (Environmental, Social, and Governance) goals is increasingly important for pharmaceutical companies seeking to minimize their carbon footprint and regulatory burden.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these isoberaline analogues. The answers are derived directly from the experimental data and technical specifications outlined in the patent documentation. Understanding these details is vital for R&D teams evaluating the feasibility of this technology for their pipeline and for procurement officers assessing vendor capabilities. The information covers aspects ranging from raw material selection to the biological efficacy of the final compounds.
Q: Why is rufloxacin chosen as the starting material for isoberaline analogues?
A: Rufloxacin is a commercially available fluoroquinolone that provides a cost-effective scaffold. Its structure allows for the retention of the hydrophilic piperazine group, which significantly improves the water solubility and bioavailability of the final indoloquinoline product compared to natural cryptolepine alkaloids.
Q: What are the advantages of using potassium borohydride over sodium borohydride in this synthesis?
A: According to the patent data, potassium borohydride is preferred because it is cheaper, less hygroscopic, and offers milder reaction conditions. This substitution enhances process stability and reduces raw material handling costs during the reductive decarboxylation step.
Q: How does this new synthetic route address drug resistance in tuberculosis?
A: The resulting isoberaline analogues combine the pharmacophores of fluoroquinolones and natural indoloquinolines. In vitro testing demonstrated that several analogues exhibit potent activity against multidrug-resistant Mycobacterium tuberculosis strains (H6, H7, H10), often outperforming standard controls like rifampicin and isoniazid.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Isoberaline Analogue Supplier
The technological breakthroughs detailed in patent CN114276369B represent a significant leap forward in anti-tuberculosis drug discovery, yet translating this potential into commercial reality requires a partner with deep technical expertise and manufacturing capacity. NINGBO INNO PHARMCHEM stands at the forefront of fine chemical manufacturing, possessing extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our state-of-the-art facilities are equipped to handle the specific reductive and cyclization chemistries required for these indoloquinoline derivatives, ensuring stringent purity specifications and rigorous QC labs validate every batch against international pharmacopoeia standards.
We invite pharmaceutical innovators and procurement leaders to collaborate with us to accelerate the development of these promising anti-TB candidates. Our team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and timeline. We encourage you to contact our technical procurement team today to request specific COA data for our reference standards and to discuss route feasibility assessments for your custom synthesis needs. Together, we can bridge the gap between laboratory innovation and life-saving medication.