Advanced Synthetic Route for Levonorgestrel Impurity O: Scalability and Commercial Viability Analysis
Advanced Synthetic Route for Levonorgestrel Impurity O: Scalability and Commercial Viability Analysis
The pharmaceutical industry continuously demands higher purity standards for reference substances to ensure the safety and efficacy of hormonal contraceptives like Levonorgestrel. A significant breakthrough in this domain is detailed in patent CN114031659A, which discloses a novel and robust preparation method for Levonorgestrel Impurity O, chemically known as 5 alpha-methoxy-17 alpha-ethynyl-17 beta-hydroxy-18-methyl-steryl-3-ketone. Historically, the synthesis of this specific impurity has been challenging, with limited documentation available in public literature, creating a bottleneck for quality control laboratories and R&D departments globally. This new technical disclosure addresses the critical need for a reliable supply of this complex steroid intermediate by outlining a pathway that utilizes cheap and easily accessible raw materials, specifically starting from Compound I (3-methoxy-18-methyl-steroid-2, 5(10)-diene-17 ketone). The innovation lies not just in the chemical transformation itself, but in the strategic selection of protecting groups and reagents that streamline the workflow, making it an attractive option for a reliable pharmaceutical intermediate supplier aiming to optimize their catalog of steroid derivatives.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior to the innovations presented in the referenced patent, the procurement of Levonorgestrel Impurity O was fraught with significant logistical and technical hurdles. The background art indicates that reference substances for Levonorgestrel related substances are notoriously difficult to purchase both domestically and internationally, primarily because the preparation methods for most of these related substances were simply not reported in existing scientific documents. This lack of transparency forced research institutions to rely on scarce commercial sources or attempt to develop their own proprietary routes, which often resulted in low yields, poor reproducibility, and excessive costs. Conventional approaches to synthesizing complex steroid impurities often involve harsh reaction conditions, expensive transition metal catalysts that require rigorous removal to meet ppm-level specifications, and multi-step purification processes that erode overall material throughput. Furthermore, the absence of a standardized, documented synthesis method meant that batch-to-batch consistency was hard to guarantee, posing a severe risk to the validation processes of pharmaceutical manufacturers who require absolute certainty in their impurity profiling.
The Novel Approach
The methodology introduced in patent CN114031659A represents a paradigm shift in how this specific steroid impurity is manufactured, offering a clear path toward cost reduction in API manufacturing. The core of this novel approach is a seven-step sequence that begins with a dual ketal protection strategy, effectively masking the reactive carbonyl functionalities at the C-3 and C-17 positions to prevent unwanted side reactions during subsequent transformations. Unlike traditional routes that might struggle with regioselectivity, this process utilizes a combination of N-halosuccinimide and magnesium oxide for halogenation and hydroxylation, followed by a clean dehalogenation step using lithium aluminum hydride. The brilliance of this route is its operational simplicity; the reagents employed are common laboratory chemicals rather than exotic catalysts, and the reaction conditions are generally mild, often proceeding at room temperature or with simple reflux. Most critically, the process allows for the direct use of obtained intermediates in the next reaction without extensive purification, a feature that dramatically reduces solvent consumption and processing time, thereby enhancing the overall economic viability of the synthesis.
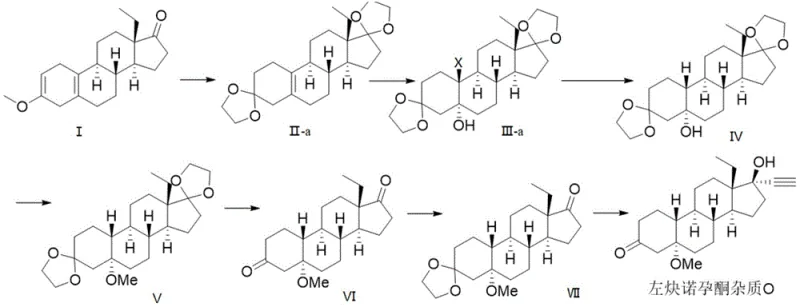
Mechanistic Insights into Steroid Functionalization and Protection Strategies
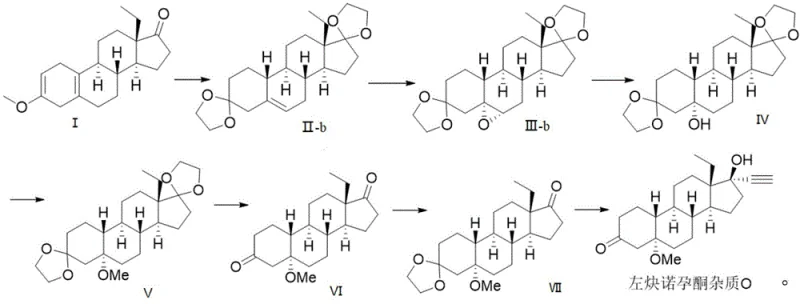
To fully appreciate the technical depth of this synthesis, one must analyze the mechanistic nuances of the functional group interconversions, particularly the strategic use of ketal protection and the subsequent ethynylation. The initial step involves reacting Compound I with triethyl orthoformate and ethylene glycol under acid catalysis, which forms a mixture of Compound II-a and a minor amount of Compound II-b. This ketalization is crucial because it stabilizes the steroid backbone against the strong nucleophiles and reducing agents used in later stages. The subsequent halogenation and hydroxylation step is particularly interesting; it proceeds via the formation of a halohydrin intermediate where the magnesium oxide acts as a mild base to facilitate the addition of the halogen species from N-halosuccinimide across the double bond. This is followed by a dehalogenation reaction using lithium aluminum hydride, which not only removes the halogen atom but also reduces the functionality to establish the correct stereochemistry at the C-5 position. The patent data confirms through TLC and NMR analysis that the hydrogen at the 10-position retains the beta configuration, verifying the high stereoselectivity of this reduction. This level of control is essential for producing a reference standard that accurately mimics the impurity profile found in the final drug product.
Furthermore, the patent discloses an alternative pathway that highlights the versatility of the intermediate compounds, specifically converting Compound II-b into the final product via an epoxidation route. In this variation, Compound II-b undergoes epoxidation using m-chloroperoxybenzoic acid to form Compound III-b, which is then subjected to a reduction ring-opening reaction. This alternative mechanism provides a valuable contingency for process optimization, allowing manufacturers to utilize the by-product (Compound II-b) from the initial protection step rather than discarding it, thus improving the atom economy of the entire process. The final stage involves a 17-position ethynylation using lithium trimethylsilylethynyl, a reagent chosen for its ability to introduce the critical alkyne moiety with high fidelity. The reaction is conducted at low temperatures (-45°C to -20°C) to control exothermicity and ensure regioselectivity, followed by a deprotection step that reveals the final ketone at the C-3 position. This meticulous attention to reaction conditions ensures that the final Levonorgestrel Impurity O is obtained with the precise structural attributes required for analytical validation.
How to Synthesize Levonorgestrel Impurity O Efficiently
Implementing this synthesis in a production environment requires strict adherence to the optimized parameters outlined in the patent to ensure maximum yield and purity. The process is designed to be telescoped where possible, meaning that the crude reaction mixtures from steps like the ketal protection or the halogenation can often be carried forward without isolation, provided that the stoichiometry is carefully managed. For R&D teams looking to replicate this, the key lies in the control of the ethynylation step, where the generation of the lithium acetylide species must be handled under inert atmosphere to prevent moisture degradation. The detailed标准化 synthesis steps see the guide below outline the specific molar ratios, such as using a mass ratio of magnesium oxide to N-halosuccinimide of approximately 1:4 to 1:15 relative to the substrate, which is critical for driving the hydroxylation to completion. By following these precise guidelines, laboratories can reliably produce high-purity steroid intermediates that meet the stringent requirements of pharmacopoeial standards.
- Perform ketal protection on Compound I at positions 3 and 17 using triethyl orthoformate and ethylene glycol to generate Compound II-a.
- Execute halogenation and hydroxylation on Compound II-a using N-halosuccinimide and magnesium oxide to form Compound III-a.
- Conduct dehalogenation with lithium aluminum hydride to yield Compound IV, followed by methyl etherification to produce Compound V.
- Remove ketal protecting groups to form Compound VI, re-protect at position 3 to get Compound VII, and finalize with 17-position ethynylation.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented synthesis route offers tangible strategic benefits that extend beyond mere chemical feasibility. The primary advantage is the substantial cost savings derived from the use of readily available starting materials and common reagents. Unlike processes that rely on precious metal catalysts or custom-synthesized building blocks, this method utilizes commodity chemicals like ethylene glycol, triethyl orthoformate, and sodium hydride, which are stable in supply and inexpensive to source in bulk. This fundamental shift in raw material strategy significantly reduces the Bill of Materials (BOM) cost, allowing for more competitive pricing structures when sourcing this critical impurity. Additionally, the ability to use intermediates directly in the next reaction step without purification translates to a drastic simplification of the manufacturing workflow. This reduction in unit operations means less equipment occupancy time, lower energy consumption for heating and cooling, and a significant decrease in solvent waste disposal costs, all of which contribute to a leaner and more sustainable production model.
- Cost Reduction in Manufacturing: The elimination of complex purification steps between intermediate stages is a major driver for cost efficiency. By avoiding column chromatography or recrystallization for every single intermediate, the process saves vast amounts of silica gel, solvents, and labor hours. This streamlined approach ensures that the overall production cost is kept low, making it economically feasible to produce this high-value reference substance at a scale that meets global demand without inflating prices. Furthermore, the high single-step yields reported in the patent examples mean that less raw material is wasted to side reactions, maximizing the output per kilogram of input and further driving down the unit cost of the final active pharmaceutical ingredient intermediate.
- Enhanced Supply Chain Reliability: Dependence on obscure or single-source reagents is a significant risk factor in pharmaceutical supply chains. This synthesis route mitigates that risk by relying on a broad base of commercially available chemicals that are produced by multiple vendors worldwide. This diversification of the supply base ensures continuity of supply even in the face of market fluctuations or geopolitical disruptions. Moreover, the robustness of the reaction conditions—many of which proceed at room temperature or standard reflux—means that the process is less sensitive to minor variations in utility supplies (such as steam or chilled water), making it easier to manufacture consistently across different facilities and ensuring a steady flow of material to downstream customers.
- Scalability and Environmental Compliance: As regulatory pressure mounts on pharmaceutical manufacturers to reduce their environmental footprint, this process offers a greener alternative to traditional steroid synthesis. The reduction in solvent usage and waste generation aligns with modern green chemistry principles, facilitating easier compliance with environmental regulations. The process is inherently scalable; the reaction kinetics and heat transfer profiles described are amenable to scale-up from kilogram to multi-ton production without requiring specialized high-pressure or cryogenic equipment (except for the final ethynylation which is manageable). This scalability ensures that suppliers can rapidly ramp up production to meet surges in demand, providing a secure and flexible supply chain partner for large-scale pharmaceutical projects.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of Levonorgestrel Impurity O based on the disclosed patent technology. These insights are derived directly from the experimental data and beneficial effects described in the documentation, providing clarity for technical buyers and quality assurance professionals. Understanding these details is crucial for integrating this intermediate into your quality control workflows and ensuring that your supply chain is resilient and compliant with international standards.
Q: What are the critical advantages of the new synthesis method for Levonorgestrel Impurity O?
A: The method described in patent CN114031659A utilizes readily available starting materials and common laboratory reagents, significantly simplifying the process compared to previous unreported methods. It allows for high single-step yields and enables intermediates to be used directly in subsequent steps without rigorous purification, drastically reducing processing time and operational complexity.
Q: How does this process ensure stereochemical control during the synthesis?
A: The process employs specific reagents like lithium aluminum hydride for dehalogenation and reduction ring-opening, which have been demonstrated to provide consistent regioselectivity and stereoselectivity. The TLC and NMR data confirm that the hydrogen at the 10-position maintains the beta configuration, ensuring the structural integrity required for the impurity reference standard.
Q: Is this synthesis route suitable for large-scale commercial production?
A: Yes, the route is designed for scalability. The use of robust reagents such as N-bromosuccinimide and the ability to telescope certain steps (using crude intermediates directly) minimizes waste and handling time. The overall yield in optimized examples reaches over 19%, which is viable for the production of high-value reference standards and intermediates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Levonorgestrel Impurity O Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-purity reference standards play in the development and quality assurance of life-saving medications. Our team of expert chemists has thoroughly analyzed the synthetic pathways described in patent CN114031659A and possesses the technical capability to execute this complex steroid synthesis with precision. We bring extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that whether you need gram quantities for method validation or kilogram scales for stability testing, we can deliver. Our state-of-the-art facilities are equipped with rigorous QC labs that employ advanced analytical techniques to verify stringent purity specifications, guaranteeing that every batch of Levonorgestrel Impurity O meets the exacting standards required by global pharmacopoeias.
We invite you to collaborate with us to optimize your supply chain for steroid intermediates. By leveraging our expertise in process chemistry, we can offer a Customized Cost-Saving Analysis tailored to your specific volume requirements, helping you identify further efficiencies in your procurement strategy. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments. Let us be your partner in ensuring the quality and consistency of your pharmaceutical products through the reliable supply of critical impurities and intermediates.