Optimized Synthesis of Zoledronic Acid Impurity B for Global Pharmaceutical Quality Control
Introduction to Advanced Impurity Control in Bisphosphonate Manufacturing
The pharmaceutical industry's relentless pursuit of therapeutic efficacy and patient safety places an immense burden on the quality control of Active Pharmaceutical Ingredients (APIs), particularly for potent drugs like Zoledronic Acid. As a third-generation bisphosphonate, Zoledronic Acid is critical for treating tumor-induced hypercalcemia and bone metastases, necessitating rigorous impurity profiling to meet global regulatory standards. The patent CN113956290A introduces a groundbreaking preparation method for Zoledronic Acid Impurity B, a critical process-related byproduct that must be meticulously monitored. This technical disclosure outlines a robust synthetic route that overcomes historical limitations in reference standard availability, utilizing a strategic combination of nucleophilic substitution and controlled phosphorylation. By optimizing reaction parameters such as solvent polarity, temperature gradients, and stoichiometric ratios, this method ensures the production of Impurity B with purity levels exceeding 98 percent. For R&D directors and quality assurance teams, access to such high-fidelity reference materials is not merely a regulatory checkbox but a fundamental requirement for validating analytical methods and ensuring batch-to-batch consistency in commercial API manufacturing.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of specific bisphosphonate impurities has been plagued by inconsistent yields and难以 control side reactions, often resulting in reference materials that lack the requisite purity for HPLC calibration. Conventional routes frequently suffer from incomplete phosphorylation or the formation of complex polymeric byproducts due to the high reactivity of phosphorus halides. Furthermore, traditional purification techniques often rely on extensive chromatography, which is neither cost-effective nor scalable for the production of kilogram quantities needed for ongoing quality control operations. The lack of a standardized, reproducible method for Impurity B has created a bottleneck in the supply chain for zoledronic acid manufacturers, forcing reliance on scarce or poorly characterized standards. This scarcity complicates the validation of cleaning procedures and the establishment of strict acceptance criteria, potentially delaying regulatory filings and market entry for generic versions of this essential bone therapy medication.
The Novel Approach
The methodology disclosed in CN113956290A represents a significant paradigm shift by introducing a streamlined, three-step sequence that prioritizes selectivity and ease of isolation. Unlike previous attempts, this novel approach leverages the specific solvation properties of sulfolane during the critical phosphorylation step to stabilize reactive intermediates and suppress side reactions. The process begins with a highly efficient nucleophilic substitution of imidazole, followed by a mild hydrolysis to generate the key acetic acid precursor. The innovation lies primarily in the final condensation step, where precise temperature control—ramping from 35°C to 65°C and finally to 95°C—allows for the sequential formation of the phosphonate bonds without degrading the sensitive imidazole ring. This thermal staging, combined with the use of phosphorous acid in a specific molar excess, drives the reaction to completion while minimizing the generation of unknown impurities, thereby simplifying the downstream purification to a simple recrystallization or pulping process.
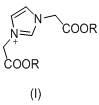
Mechanistic Insights into Phosphorylation and Impurity Formation
The core chemical transformation in this synthesis involves the conversion of (1H-imidazol-1-yl)acetic acid (Intermediate II) into the diphosphonic acid derivative through a reaction with phosphorus trichloride and phosphorous acid. Mechanistically, this proceeds via the formation of a reactive phosphite intermediate which subsequently undergoes condensation. The choice of sulfolane as the solvent is critical; its high boiling point and polar aprotic nature facilitate the dissolution of both the organic acid and the inorganic phosphorus species, creating a homogeneous reaction environment that enhances mass transfer. The initial stirring at 35 +/- 5°C allows for the formation of a stable complex between the carboxylic acid and the phosphorus reagents before the exothermic addition of phosphorus trichloride. Subsequent heating to 65 +/- 5°C promotes the nucleophilic attack of the phosphorus species onto the alpha-carbon, while the final hydrolysis step at 95 +/- 5°C ensures the complete conversion of any P-Cl bonds to P-OH groups, yielding the thermodynamically stable diphosphonic acid structure characteristic of Impurity B.
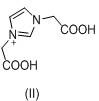
Controlling the impurity profile during this synthesis requires a deep understanding of the competing reaction pathways. The patent specifies a molar ratio of Intermediate II to phosphorous acid of 1:4 to 6, which is significantly higher than stoichiometric requirements. This excess serves a dual purpose: it acts as a scavenger for moisture that could prematurely hydrolyze the phosphorus trichloride, and it drives the equilibrium towards the desired bis-phosphonated product by Le Chatelier's principle. Furthermore, the slow addition of water post-reaction at room temperature, followed by heating, prevents the violent exotherm typically associated with phosphorus halide hydrolysis, which can otherwise lead to charring or decomposition of the organic backbone. By maintaining the reaction system below 60°C before the final hydrolysis ramp, the process effectively mitigates the risk of forming cyclic phosphate esters or other thermal degradation products, ensuring that the final crude product is already of high purity before the final methanol pulping step.
How to Synthesize Zoledronic Acid Impurity B Efficiently
Implementing this synthesis route in a pilot or production facility requires strict adherence to the thermal profiles and stoichiometric ratios defined in the patent examples to guarantee reproducibility. The process is designed to be operationally simple, avoiding the need for cryogenic conditions or anhydrous glovebox techniques, which significantly lowers the barrier to entry for contract manufacturing organizations. The initial alkylation of imidazole can be performed in standard glass-lined reactors using dichloromethane or alternative green solvents, followed by a straightforward aqueous workup. The critical phosphorylation step demands precise temperature monitoring equipment to maintain the specific 35°C to 65°C window, ensuring the reaction kinetics favor the formation of Impurity B over potential mono-phosphonated side products. Detailed standardized operating procedures (SOPs) regarding the addition rate of phosphorus trichloride and the subsequent water quench are essential to manage gas evolution and thermal safety. For a comprehensive breakdown of the exact reagent quantities, reaction times, and workup protocols, please refer to the step-by-step guide below.
- Perform nucleophilic substitution of imidazole with haloacetate (e.g., methyl chloroacetate) using potassium carbonate and sodium iodide catalyst in dichloromethane at 20-40°C for 24 hours to form Intermediate I.
- Hydrolyze Intermediate I by adding water and heating to 70-80°C for 10 hours, followed by dispersion in ethanol and purification to obtain Intermediate II ((1H-imidazol-1-yl)acetic acid).
- React Intermediate II with phosphorous acid and phosphorus trichloride in sulfolane solvent, controlling temperature stages at 35°C, 65°C, and finally 95°C after water addition to yield the final impurity.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, the adoption of this optimized synthesis route offers substantial strategic benefits, primarily driven by the simplification of the manufacturing process and the use of commodity chemicals. The elimination of complex catalytic systems or rare earth metals means that the raw material supply chain is robust and resistant to geopolitical fluctuations or shortages. The reliance on bulk solvents like sulfolane, methanol, and dichloromethane ensures that production costs remain predictable and low, facilitating significant cost reduction in pharmaceutical intermediate manufacturing. Moreover, the high crude purity achieved directly from the reaction minimizes the need for expensive preparative HPLC purification, which is traditionally the most costly step in reference standard production. This efficiency translates directly into shorter lead times for high-purity pharmaceutical intermediates, allowing suppliers to respond more rapidly to the urgent quality control needs of API manufacturers during regulatory audits or batch release testing.
- Cost Reduction in Manufacturing: The process achieves cost optimization by utilizing inexpensive, widely available starting materials such as imidazole and chloroacetic acid derivatives, avoiding the need for custom-synthesized precursors. The high selectivity of the reaction reduces the consumption of solvents and adsorbents typically required for extensive purification, leading to lower waste disposal costs and improved overall process economics. By streamlining the workflow into three distinct, high-yielding steps, the labor hours and reactor occupancy time are significantly reduced, enhancing the throughput capacity of existing manufacturing facilities without the need for capital-intensive equipment upgrades.
- Enhanced Supply Chain Reliability: The robustness of this synthetic route ensures a consistent supply of Zoledronic Acid Impurity B, mitigating the risk of stockouts that can halt API production lines. Since the method does not depend on single-source catalysts or proprietary reagents, procurement teams can diversify their supplier base for raw materials, increasing resilience against supply chain disruptions. The scalability of the process from gram to kilogram scales means that suppliers can easily ramp up production volumes to meet surging demand from generic drug manufacturers entering the market, ensuring long-term continuity of supply for critical quality control materials.
- Scalability and Environmental Compliance: The process is inherently scalable, with reaction conditions that are easily managed in large-scale industrial reactors, supporting the commercial scale-up of complex pharmaceutical intermediates. The use of recyclable solvents and the generation of manageable inorganic byproducts align with modern green chemistry principles, simplifying environmental compliance and waste treatment protocols. This environmental compatibility reduces the regulatory burden on manufacturing sites and supports the sustainability goals of multinational pharmaceutical corporations seeking to minimize the carbon footprint of their supply chains.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of Zoledronic Acid Impurity B. These insights are derived directly from the experimental data and process descriptions found in the underlying patent literature, providing a reliable foundation for decision-making. Understanding the nuances of this synthesis helps stakeholders appreciate the value of high-quality reference standards in maintaining drug safety and efficacy. For further technical specifications or custom synthesis requests, our team is prepared to provide detailed documentation and support.
Q: What is the primary advantage of this synthesis method for Zoledronic Acid Impurity B?
A: The primary advantage is the ability to achieve purity levels exceeding 98% through optimized solvent selection (sulfolane) and precise temperature staging during the phosphorylation step, making it ideal for use as a qualified reference substance.
Q: Why is controlling Impurity B critical in Zoledronic Acid manufacturing?
A: Impurity B is a key process-related impurity generated during the synthesis of Zoledronic Acid. Strict control and quantification using high-purity reference standards are mandatory to ensure the safety, efficacy, and regulatory compliance of the final bisphosphonate drug product.
Q: Can this process be scaled for commercial production of reference materials?
A: Yes, the process utilizes common industrial reagents like imidazole, chloroacetates, and phosphorus trichloride, and avoids exotic catalysts, making it highly amenable to scale-up from gram-scale laboratory synthesis to multi-kilogram commercial production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Zoledronic Acid Impurity B Supplier
At NINGBO INNO PHARMCHEM, we recognize that the integrity of your final drug product depends on the quality of every component in your supply chain, including the reference standards used for quality control. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet your volume requirements with consistent quality. We employ stringent purity specifications and operate rigorous QC labs equipped with state-of-the-art analytical instrumentation to verify that every batch of Zoledronic Acid Impurity B meets the >98% purity benchmark required for regulatory submissions. Our commitment to technical excellence means that we do not just supply chemicals; we provide solutions that de-risk your development timeline and ensure compliance with global pharmacopoeia standards.
We invite you to collaborate with us to optimize your impurity management strategy and secure a stable supply of critical reference materials. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific production volumes. We are ready to provide specific COA data, route feasibility assessments, and competitive pricing models that demonstrate the tangible value of partnering with a leader in fine chemical manufacturing. Let us support your journey towards delivering safe and effective therapies to patients worldwide.