Advanced Synthesis of 2-Cyanoacetyl-5-Substituted Thiophenes for High-Purity Pharmaceutical Manufacturing
Advanced Synthesis of 2-Cyanoacetyl-5-Substituted Thiophenes for High-Purity Pharmaceutical Manufacturing
The pharmaceutical industry constantly seeks robust synthetic routes for heterocyclic intermediates that serve as the backbone for novel therapeutic agents. Patent CN101397291A introduces a significant technological advancement in the preparation of 2-cyanoacetyl-5-substituted thiophene compounds, which are critical precursors for potential anti-arthritic drugs. This proprietary methodology addresses long-standing challenges in heterocyclic chemistry, specifically regarding the stability of halogenated substituents during acylation reactions. By leveraging a precise two-step sequence involving iodine-mediated oxidation followed by cryogenic nucleophilic substitution, the process achieves exceptional chemical fidelity. For R&D directors and procurement specialists seeking a reliable pharmaceutical intermediate supplier, understanding the nuances of this patent is essential for securing high-quality raw materials. The technology not only ensures superior product consistency but also aligns with modern green chemistry principles by optimizing reagent usage and minimizing hazardous waste streams.
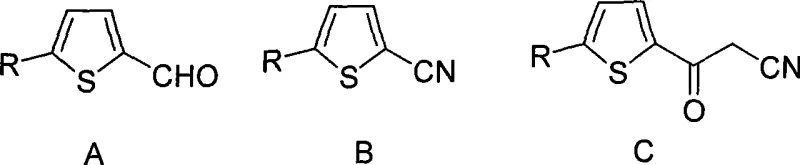
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 2-cyanoacetyl-5-halogenated thiophenes has been plagued by significant chemoselectivity issues, particularly when dealing with sensitive iodine substituents. Prior art, such as the methods reported by Ridge et al., typically relied on strong bases like sodium hydride to facilitate the condensation of thiophene carbonitriles with acetonitrile. However, these aggressive conditions often triggered unwanted side reactions, most notably the partial deiodination of the starting material. When the substituent R is iodine, the use of sodium hydride frequently resulted in a complex mixture of the desired 2-cyanoacetyl-5-iodothiophene and the dehalogenated by-product, 2-cyanoacetylthiophene. Since these two compounds exhibit remarkably similar solubility profiles in common organic solvents, separating them through standard crystallization or chromatography becomes an arduous and yield-depleting task. This inability to effectively control impurity profiles rendered previous methods unsuitable for the rigorous purity standards demanded by the pharmaceutical sector, often necessitating costly and time-consuming downstream purification processes that eroded overall process efficiency.
The Novel Approach
The methodology disclosed in CN101397291A represents a paradigm shift by decoupling the oxidation and acylation steps into highly controlled distinct phases. Instead of relying on harsh hydride bases at ambient or elevated temperatures, this novel approach utilizes a carefully tuned low-temperature regime ranging from -80°C to 0°C during the critical acylation step. By employing organolithium reagents such as n-butyllithium or alkoxides like sodium methoxide under these cryogenic conditions, the reaction kinetics are modulated to favor nucleophilic attack on the nitrile group while suppressing competitive dehalogenation pathways. Furthermore, the initial oxidation step converts readily available 5-substituted thiophene-2-carbaldehydes into the corresponding nitriles using an iodine-ammonia water system, which is both atom-economical and operationally simple. This strategic modification of reaction parameters allows for the successful synthesis of previously difficult-to-access iodo-substituted derivatives with high regioselectivity. Consequently, the final products are obtained with minimal by-product formation, drastically simplifying the isolation procedure and ensuring that the material meets the stringent specifications required for drug development pipelines.
Mechanistic Insights into Iodine-Mediated Oxidation and Cryogenic Acylation
The first stage of this synthesis involves the transformation of a 5-substituted thiophene-2-carbaldehyde into a 5-substituted thiophenecarbonitrile using molecular iodine and aqueous ammonia in an inert organic solvent. Mechanistically, this likely proceeds through the formation of an imine intermediate followed by oxidative dehydration, where iodine acts as the oxidant to drive the equilibrium toward the nitrile product. The choice of solvent is critical here; inert organic solvents such as dioxane, ethers, or nitriles provide the necessary medium to solubilize the organic aldehyde while allowing effective interaction with the aqueous ammonia phase. The reaction temperature is maintained between 0°C and 60°C, a range that provides sufficient thermal energy to overcome the activation barrier for oxidation without promoting the degradation of the sensitive thiophene ring or the halogen substituents. Following the reaction, the addition of sodium thiosulfate effectively quenches any unreacted iodine, preventing potential halogenation side reactions on the thiophene ring and ensuring a clean crude product that can be easily isolated via cooling and crystallization.
In the second mechanistic phase, the generated nitrile undergoes nucleophilic addition with acetonitrile anions. This step requires the generation of a highly reactive acetonitrile carbanion, typically achieved by deprotonating acetonitrile with a strong base like n-butyllithium or sodium amide at temperatures as low as -80°C. The extreme cold is not merely a precaution but a fundamental requirement to stabilize the lithiated intermediate and prevent it from acting as a base towards the aromatic protons or the carbon-halogen bond, which would lead to decomposition. Once the nucleophile is formed, the 5-substituted thiophenecarbonitrile is added dropwise, allowing for a controlled reaction that minimizes local exotherms and concentration gradients. The resulting imine anion is subsequently hydrolyzed upon acidic workup to yield the stable 2-cyanoacetyl ketone. This precise control over the reaction environment is the key differentiator that enables the preservation of labile groups like iodine, thereby expanding the chemical space available for medicinal chemists to explore structure-activity relationships in anti-arthritic drug candidates.
How to Synthesize 2-Cyanoacetyl-5-Substituted Thiophenes Efficiently
Implementing this synthesis route in a laboratory or pilot plant setting requires strict adherence to the temperature profiles and reagent stoichiometry outlined in the patent data to ensure reproducibility and safety. The process begins with the preparation of the nitrile intermediate, where maintaining the correct molar ratio of iodine to aldehyde (preferably 1.5 to 2 equivalents) is crucial for driving the reaction to completion without excessive waste. Operators must monitor the reaction progress via HPLC to determine the optimal endpoint before quenching with sodium thiosulfate and inducing crystallization by cooling the mixture to 0°C. The subsequent acylation step demands even greater precision, requiring the use of dry, oxygen-free inert solvents like tetrahydrofuran or benzene and the maintenance of cryogenic temperatures throughout the addition of the base and the nitrile substrate. Detailed standardized operating procedures for these sensitive low-temperature reactions are essential for training technical staff and ensuring consistent batch-to-batch quality.
- Oxidize 5-substituted thiophene-2-carbaldehyde with iodine and ammonia water in an inert organic solvent at 0 to 60°C to form the nitrile intermediate.
- React the resulting 5-substituted thiophenecarbonitrile with acetonitrile in the presence of a strong base at cryogenic temperatures (-80 to 0°C).
- Quench the reaction with acid, extract with organic solvents, and purify via crystallization to obtain the final 2-cyanoacetyl product with >98% purity.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this patented synthesis route offers substantial strategic advantages for procurement managers and supply chain leaders focused on cost reduction in pharmaceutical intermediate manufacturing. The primary economic driver is the significant simplification of the purification process; because the reaction selectively avoids the formation of difficult-to-remove dehalogenated by-products, the need for extensive chromatographic purification is eliminated. This reduction in downstream processing directly translates to lower solvent consumption, reduced labor hours, and decreased waste disposal costs, all of which contribute to a more favorable cost of goods sold (COGS). Furthermore, the reliance on commodity chemicals such as iodine, ammonia water, and acetonitrile ensures that the raw material supply chain is robust and less susceptible to the volatility often associated with specialized catalytic reagents. This stability in sourcing allows for more accurate long-term budgeting and reduces the risk of production delays caused by material shortages.
- Cost Reduction in Manufacturing: The elimination of expensive transition metal catalysts and the avoidance of complex separation techniques for isomeric impurities create a leaner manufacturing process. By utilizing straightforward crystallization as the primary purification method, the process minimizes the loss of valuable material that often occurs during column chromatography, thereby improving the overall effective yield. Additionally, the ability to recycle solvents such as ethers and nitriles further enhances the economic viability of the process, making it an attractive option for large-volume production where marginal savings per kilogram compound into significant financial benefits.
- Enhanced Supply Chain Reliability: The use of widely available, non-proprietary reagents means that the supply chain is not dependent on single-source vendors for exotic catalysts. This diversification of supply sources mitigates the risk of disruptions and ensures continuity of supply, which is critical for maintaining the production schedules of downstream API manufacturers. The operational simplicity of the method also means that it can be easily transferred between different manufacturing sites or contract manufacturing organizations (CMOs) without the need for highly specialized equipment, providing greater flexibility in capacity planning and inventory management.
- Scalability and Environmental Compliance: The process is inherently designed for scale-up, utilizing standard unit operations like stirred tank reactors and filtration units that are commonplace in fine chemical facilities. The absence of heavy metals in the reaction scheme simplifies the environmental compliance profile, reducing the burden of wastewater treatment and residual metal testing in the final product. This alignment with green chemistry principles not only lowers regulatory hurdles but also appeals to increasingly eco-conscious stakeholders, positioning the manufacturer as a responsible partner in the sustainable development of pharmaceutical supply chains.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology, derived directly from the patent specifications and practical application scenarios. Understanding these details helps stakeholders evaluate the feasibility of integrating this intermediate into their specific drug development programs. The answers reflect the high standards of purity and process control that define this manufacturing approach, ensuring transparency for all partners involved in the value chain.
Q: How does this method prevent deiodination in iodo-substituted thiophenes?
A: Unlike conventional methods using sodium hydride which cause partial deiodination, this patent utilizes controlled low-temperature conditions (-80 to 0°C) and specific organolithium or alkoxide bases to preserve the iodine substituent while facilitating acylation.
Q: What is the achievable purity for these pharmaceutical intermediates?
A: The described process consistently yields products with HPLC purity exceeding 98%, meeting the stringent requirements for scientific research and pharmaceutical industry applications without complex purification steps.
Q: Is this synthesis route suitable for large-scale industrial production?
A: Yes, the method employs commercially available reagents like iodine, ammonia water, and acetonitrile, and utilizes standard unit operations such as crystallization and extraction, making it highly scalable for commercial manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Cyanoacetyl-5-Substituted Thiophenes Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality heterocyclic intermediates play in the discovery and development of next-generation therapeutics. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to industrial reactor is seamless and efficient. We are committed to delivering 2-cyanoacetyl-5-substituted thiophenes with stringent purity specifications, leveraging our rigorous QC labs to verify that every batch meets the >98% HPLC purity benchmark required for pharmaceutical applications. Our state-of-the-art facilities are equipped to handle the cryogenic conditions and inert atmosphere requirements of this synthesis, guaranteeing product integrity and consistency.
We invite procurement leaders and R&D directors to collaborate with us to optimize their supply chains for anti-arthritic drug candidates. By partnering with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. We encourage you to reach out today to obtain specific COA data and route feasibility assessments that demonstrate how our advanced manufacturing capabilities can support your project timelines and budgetary goals. Let us be your trusted partner in bringing innovative medicines to market faster and more efficiently.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →