Advanced Synthesis of High-Purity Fulvestrant Related Substance IV for Global Quality Control Standards
Advanced Synthesis of High-Purity Fulvestrant Related Substance IV for Global Quality Control Standards
The pharmaceutical industry's relentless pursuit of oncology therapeutics has placed Fulvestrant, a potent estrogen receptor downregulator, at the forefront of breast cancer treatment protocols. However, the rigorous quality control required for such potent active pharmaceutical ingredients (APIs) necessitates the availability of highly pure related substances for accurate impurity profiling. Patent CN114671922A introduces a groundbreaking preparation method for Fulvestrant Related Substance IV, addressing critical gaps in the current supply of reference standards. This innovation provides a robust synthetic pathway that bypasses the hazardous and inefficient conditions of legacy processes, delivering a reference material with exceptional purity suitable for both qualitative and quantitative analysis in GMP environments. By leveraging mild reaction conditions and avoiding complex purification techniques, this technology represents a significant leap forward for reliable pharmaceutical intermediate suppliers aiming to support global drug safety initiatives.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
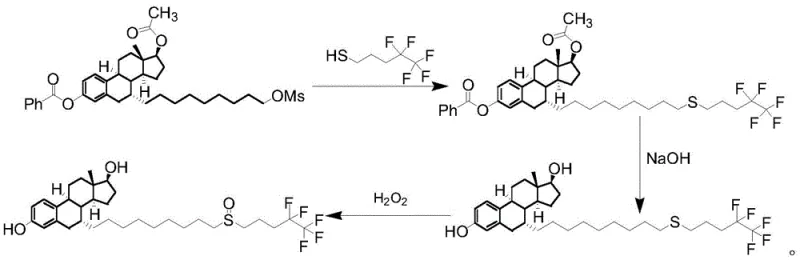
Historically, the synthesis of Fulvestrant and its associated impurities has been plagued by significant operational and safety challenges that hinder scalable manufacturing. Early methodologies, such as those described in Patent US4659516, relied heavily on pentafluoro-pentanethiol, a reagent notorious for its extreme sensitivity to air, difficult synthesis, exorbitant cost, and intensely pungent odor, rendering it unsuitable for large-scale industrial production. Furthermore, alternative routes like those in Patent WO2003031399 involved harsh reaction conditions and hydrogenation steps that posed substantial safety risks during scale-up while suffering from low yields and poor product quality. Perhaps most critically, processes outlined in Patent US20060030552, while utilizing more accessible raw materials, were prone to generating persistent disulfide impurities and unwanted nucleophilic substitutions at the 3-position phenolic hydroxyl group. These legacy methods often necessitated multiple rounds of column chromatography to achieve acceptable purity, a technique that is economically prohibitive and technically difficult to translate from the laboratory to commercial manufacturing scales.
The Novel Approach
In stark contrast to these cumbersome legacy techniques, the novel approach detailed in CN114671922A utilizes a sophisticated two-step strategy that prioritizes safety, efficiency, and purity. This method replaces the hazardous thiol reagents with stable pentafluoropentyl sulfonates, facilitating a clean nucleophilic substitution under mild alkaline conditions. The process is designed to minimize side reactions, effectively suppressing the formation of disulfide by-products and ensuring that the phenolic hydroxyl group reacts selectively. By optimizing the reaction parameters, including temperature control and solvent selection, the synthesis achieves high conversion rates for each intermediate without the need for dangerous hydrogenation or complex workup procedures. The culmination of this streamlined process is a crude product that can be purified to reference-grade standards through simple recrystallization, completely eliminating the need for resource-intensive column chromatography and significantly enhancing the feasibility of commercial scale-up of complex pharmaceutical intermediates.
Mechanistic Insights into Controlled Nucleophilic Substitution and Selective Oxidation
The core of this technological advancement lies in the precise orchestration of a nucleophilic substitution reaction followed by a highly selective oxidation step. In the first stage, a specialized steroid precursor (Compound I) reacts with a fluorinated sulfonate ester (Compound II) in a polar aprotic solvent system such as DMF or DMAC. The presence of a controlled alkaline environment, typically provided by sodium or potassium hydroxide solutions, deprotonates the phenolic hydroxyl group, activating it for nucleophilic attack on the sulfonate leaving group. This step is meticulously managed at temperatures between 0°C and 30°C during reagent addition to prevent thermal runaway, followed by a heat preservation phase at 30°C to 60°C to drive the reaction to completion. This careful thermal management ensures that the ether linkage is formed exclusively at the desired position without compromising the integrity of the sensitive steroid backbone or the fluorinated side chains.
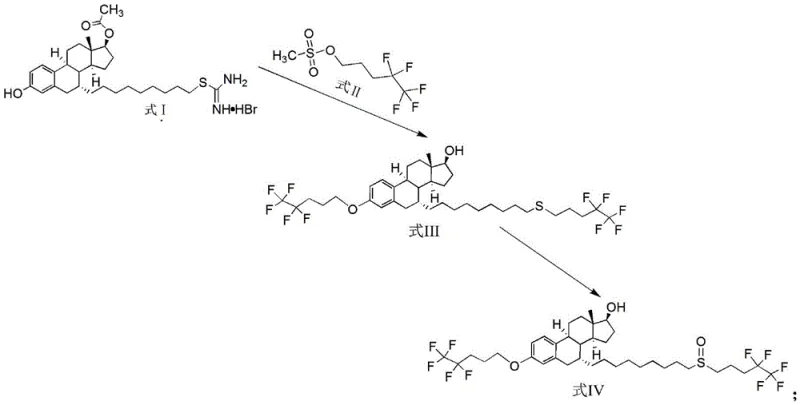
Following the formation of the sulfide intermediate (Compound III), the process transitions to a critical oxidation phase to generate the target sulfoxide functionality found in Related Substance IV. This transformation is achieved using hydrogen peroxide in the presence of glacial acetic acid, which acts as a promoter for the selective oxidation of the sulfur atom. The reaction is conducted under strict temperature control, typically between 10°C and 25°C, to prevent over-oxidation to the sulfone, a common impurity that is difficult to separate. The stoichiometry is carefully balanced, with a molar ratio of hydrogen peroxide to substrate maintained around 1:2.2, ensuring complete conversion while minimizing oxidative stress on the molecule. The resulting product exhibits exceptional stability and purity, with the mechanistic pathway inherently suppressing the formation of the disulfide and phenolic substitution impurities that plague conventional methods, thereby securing a high-quality profile for high-purity pharmaceutical intermediates.
How to Synthesize Fulvestrant Related Substance IV Efficiently
The synthesis of this critical reference standard is designed for operational simplicity and reproducibility, making it an ideal candidate for technology transfer to manufacturing facilities. The procedure begins with the dissolution of the steroid precursor and the fluorinated sulfonate in a suitable organic solvent, followed by the controlled addition of an aqueous alkali solution to initiate the coupling reaction. After the initial substitution and hydrolysis are complete, the intermediate is isolated and subjected to the oxidation protocol using hydrogen peroxide under acidic conditions. The entire workflow is optimized to maximize yield and minimize waste, with the final purification achieved through a straightforward recrystallization from ethyl acetate and n-heptane. For laboratory and pilot plant teams looking to implement this methodology, the detailed standardized synthesis steps are provided below to ensure consistent results.
- Perform nucleophilic substitution and hydrolysis between Compound I and Pentafluoropentyl Sulfonate (Compound II) in an alkaline organic solvent environment to generate Compound III.
- Oxidize Compound III using hydrogen peroxide in a solvent system with glacial acetic acid under controlled low temperatures to form the target sulfoxide, Related Substance IV.
- Execute post-treatment via liquid separation, washing, and final recrystallization using ethyl acetate and n-heptane to achieve purity levels exceeding 99.8%.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this novel synthesis route offers transformative benefits that extend far beyond simple chemical efficacy. By eliminating the reliance on malodorous and hazardous thiol reagents, the process drastically reduces the complexity of environmental health and safety (EHS) compliance, lowering the barrier for entry for contract manufacturing organizations. The removal of column chromatography from the purification train is a particularly significant economic driver, as it replaces a batch-oriented, solvent-intensive bottleneck with a continuous-friendly crystallization process. This shift not only reduces solvent consumption and waste disposal costs but also significantly shortens the production cycle time, allowing for faster response to market demands for quality control standards. Consequently, this technology enables cost reduction in pharmaceutical intermediate manufacturing by streamlining operations and reducing the dependency on expensive purification media.
- Cost Reduction in Manufacturing: The elimination of column chromatography represents a massive reduction in operational expenditure, as silica gel and large volumes of elution solvents are no longer required for purification. Furthermore, the use of stable, commercially available sulfonate esters instead of custom-synthesized thiols reduces raw material costs and minimizes the risk of batch failures due to reagent instability. The high yield and purity of the intermediates mean that less material is lost to side reactions, maximizing the return on investment for every kilogram of starting material processed.
- Enhanced Supply Chain Reliability: By avoiding reagents that are sensitive to air and difficult to store, the supply chain becomes more resilient and less prone to disruptions caused by reagent degradation. The mild reaction conditions allow for the use of standard stainless steel reactors without the need for specialized lining or exotic metallurgy required for handling corrosive or hazardous thiols. This compatibility with standard equipment ensures that multiple qualified manufacturers can produce the material, reducing single-source risk and ensuring a steady supply of high-purity pharmaceutical intermediates for global quality control laboratories.
- Scalability and Environmental Compliance: The transition from chromatography to recrystallization is a key enabler for industrial scale-up, as crystallization is a unit operation that scales linearly and predictably from grams to tons. The reduced solvent load and the absence of silica waste significantly lower the environmental footprint of the manufacturing process, aligning with green chemistry principles and stringent regulatory requirements. This scalability ensures that reducing lead time for high-purity pharmaceutical intermediates is achievable without compromising on the rigorous purity specifications demanded by regulatory bodies.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of Fulvestrant Related Substance IV. These insights are derived directly from the patent data to provide clarity on the process advantages and quality attributes. Understanding these details is crucial for stakeholders evaluating the integration of this reference standard into their quality assurance workflows.
Q: Why is this new synthesis route superior to prior art methods like US4659516?
A: Unlike prior art which utilizes sensitive, pungent, and expensive pentafluoro-pentanethiol, this novel method employs stable sulfonate esters and mild alkaline conditions, eliminating safety hazards and simplifying industrial handling.
Q: How does this process improve impurity control for Fulvestrant manufacturing?
A: The method specifically targets the formation of Related Substance IV with high selectivity, avoiding the accumulation of disulfide impurities and nucleophilic substitution by-products common in older routes, thus enabling simpler purification via recrystallization rather than column chromatography.
Q: What are the typical purity levels achievable with this reference standard synthesis?
A: Experimental data from the patent indicates that the final product can consistently achieve HPLC purity levels greater than 99.8% after simple recrystallization, making it ideal for use as a qualified reference substance.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Fulvestrant Related Substance IV Supplier
At NINGBO INNO PHARMCHEM, we recognize that the integrity of your final drug product depends on the quality of the reference standards used to validate it. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the sophisticated chemistry described in CN114671922A can be translated into a robust, GMP-compliant manufacturing process. We are committed to meeting stringent purity specifications through our rigorous QC labs, guaranteeing that every batch of Fulvestrant Related Substance IV meets the exacting standards required for global regulatory submissions. Our infrastructure is designed to handle complex fluorinated chemistries safely and efficiently, providing a secure source for your critical quality control needs.
We invite you to collaborate with us to optimize your supply chain for oncology reference standards. By engaging with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage potential partners to contact us to obtain specific COA data and route feasibility assessments, ensuring that our capabilities align perfectly with your project timelines and quality objectives. Let us be your partner in delivering safer, higher-quality medicines to patients worldwide.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →