Advanced Manufacturing of Substituted Nucleoside Analogs for Antiviral Therapeutics
Advanced Manufacturing of Substituted Nucleoside Analogs for Antiviral Therapeutics
The pharmaceutical industry continuously seeks robust and scalable synthetic routes for complex active pharmaceutical ingredients (APIs), particularly in the realm of antiviral therapeutics. Patent CN107108681B discloses a groundbreaking methodology for the preparation of substituted nucleoside analogs and their intermediates, specifically targeting diseases such as viral infections including Respiratory Syncytial Virus (RSV). This technical disclosure represents a significant leap forward in process chemistry, addressing critical bottlenecks related to impurity control, yield optimization, and scalability that have historically plagued the manufacturing of nucleoside-based drugs. By leveraging a novel sequence of protection, oxidation, cyclization, and functionalization steps, the inventors have established a pathway that not only ensures high chemical purity but also facilitates commercial-scale production without the reliance on resource-intensive chromatographic separations.
For R&D directors and process chemists, the value of this patent lies in its meticulous attention to stereochemical integrity and impurity management. The synthesis begins with readily available starting materials and proceeds through a series of well-defined transformations, utilizing reagents such as triisopropylsilyl chloride for protection and trifluoromethanesulfonic anhydride for activation. The strategic design of this route allows for the precise installation of functional groups at the 4' and 5' positions of the ribose ring, which is crucial for the biological activity of the final nucleoside analog. Furthermore, the process demonstrates exceptional reproducibility when transitioning from gram-scale laboratory experiments to kilogram-scale manufacturing, a feat that is often difficult to achieve in nucleoside chemistry due to the sensitivity of intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthetic routes for nucleoside analogs frequently suffer from a myriad of technical and economic disadvantages that hinder their viability for large-scale commercial production. A primary concern in conventional methodologies is the heavy reliance on column chromatography for the purification of intermediates, which is not only cost-prohibitive at the kilogram or ton scale but also introduces significant delays in production timelines. Additionally, older processes often struggle with poor impurity profiles, where structurally similar by-products are difficult to separate from the desired product, leading to final API purities that may fall short of stringent regulatory requirements. The use of harsh reaction conditions or unstable intermediates in traditional routes can also result in low overall yields and inconsistent batch-to-batch quality, creating substantial risks for supply chain continuity.
Moreover, conventional methods often fail to optimize the physical properties of the final compound, such as crystallinity and bulk density, which are critical for downstream formulation and tablet compression. The inability to control polymorphism or achieve high bulk density can lead to poor flowability and handling issues during the drug product manufacturing phase. These limitations collectively increase the cost of goods sold (COGS) and extend the time-to-market for new antiviral therapies, making it imperative for pharmaceutical companies to adopt more efficient and robust synthetic strategies that can overcome these inherent process deficiencies.
The Novel Approach
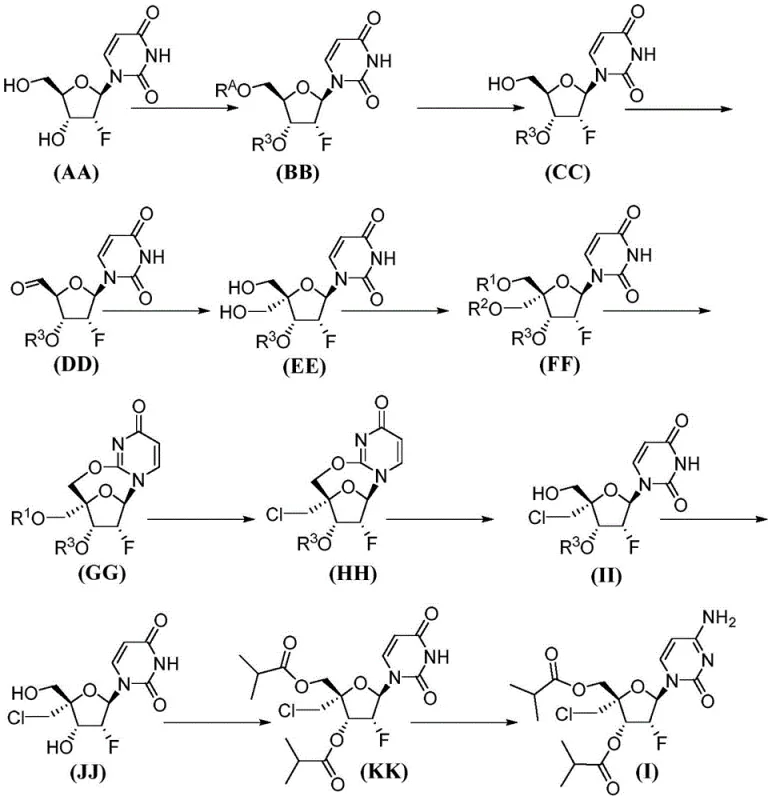
The novel approach detailed in the patent data offers a transformative solution to these challenges by introducing a streamlined synthesis that eliminates the need for chromatographic purification of intermediates. This is achieved through a carefully orchestrated sequence of reactions that favor the formation of crystalline solids at key stages, allowing for purification via simple filtration and washing. For instance, the conversion of compound (EE) to compound (FF) via bis-triflation, followed by cyclization to compound (GG), is designed to minimize side reactions and maximize the recovery of the desired stereoisomer. The use of specific reagents like DBU for cyclization and LiCl for chlorination ensures high selectivity and yield, thereby reducing the burden on downstream purification steps.
Furthermore, this innovative route significantly enhances the purity profile of the final product, Compound (I). Data indicates that impurities which are prevalent in small-scale syntheses are either completely eliminated or reduced by factors ranging from 1.5 to 10 times in the optimized large-scale process. The final product achieves a purity of up to 99.9%, surpassing the standards typically met by conventional methods. This improvement is not merely a statistical gain but a fundamental enhancement in the chemical quality of the API, ensuring greater safety and efficacy for patients. The process also improves the bulk density and crystallinity of the compound, facilitating easier handling and formulation, which translates to tangible operational efficiencies for pharmaceutical manufacturers.
Mechanistic Insights into Cyclization and Functionalization Strategies
A deep dive into the reaction mechanism reveals the sophistication of the cyclization and functionalization steps that define this synthetic route. The transformation of compound (FF) to compound (GG) involves a base-catalyzed intramolecular cyclization that forms a 7-membered ring between the uracil base and the 5'-oxygen. This step is critical as it establishes the core scaffold of the nucleoside analog with high stereocontrol. The use of amidine reagents such as DBU or DBN as bases promotes this cyclization efficiently in polar aprotic solvents, minimizing the formation of open-chain by-products. The subsequent chlorination of compound (GG) to form compound (HH) utilizes lithium chloride in a DME-DMPU solvent system, a choice that optimizes the nucleophilic substitution at the 4' position while preserving the integrity of the sensitive glycosidic bond.
The final stages of the synthesis involve the hydrolysis of the 7-membered ring to regenerate the uracil base in compound (II), followed by deprotection and acylation to yield compound (KK). The conversion of the uracil moiety to cytosine in the final step is achieved through sulfonation followed by ammonolysis, a classic yet highly effective transformation for installing the exocyclic amine. The use of 2,4,6-triisopropylbenzenesulfonyl chloride as the sulfonating agent ensures high reactivity and selectivity, while the subsequent treatment with ammonium hydroxide cleanly affords the target cytosine analog. This mechanistic pathway is robust against variations in reaction conditions, making it highly suitable for industrial scale-up where precise control over every parameter is challenging.
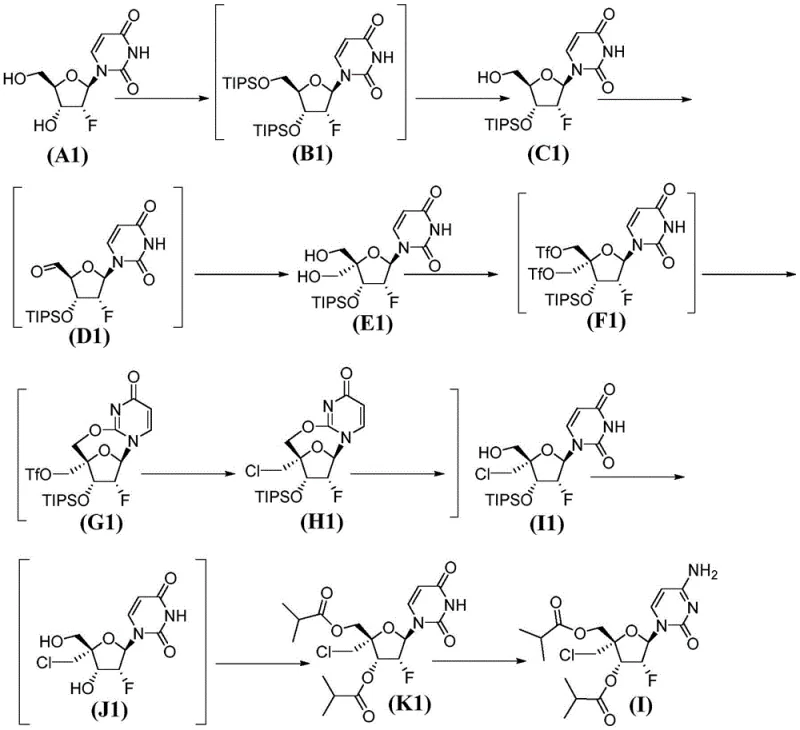
Impurity control is embedded within the mechanism itself. For example, the specific choice of protecting groups (TIPS) and the order of deprotection steps prevent the migration of acyl groups or the formation of regioisomers that are common pitfalls in nucleoside synthesis. The oxidation step using oxalyl chloride and DMSO (Swern conditions) is performed at low temperatures to prevent over-oxidation or degradation of the sugar moiety. By understanding these mechanistic nuances, process chemists can further optimize reaction parameters such as temperature, stoichiometry, and addition rates to push the purity profile even higher, ensuring that the final API meets the most rigorous pharmacopeial standards.
How to Synthesize Substituted Nucleoside Analog Efficiently
The synthesis of Compound (I) described in the patent provides a clear roadmap for manufacturing high-quality nucleoside analogs. The process begins with the dual protection of the starting nucleoside, followed by a sequence of oxidation, condensation, and reduction to modify the sugar ring. Key to the success of this route is the formation of the bis-triflate intermediate and its subsequent cyclization, which locks in the desired stereochemistry. The detailed experimental procedures provided in the patent examples demonstrate the feasibility of executing these steps on a multi-kilogram scale, with specific attention paid to temperature control and workup procedures to ensure safety and yield.
- Protect the 5'-OH and 3'-OH groups of the starting nucleoside using silyl protecting groups such as triisopropylsilyl (TIPS) to form compound (BB).
- Perform selective deprotection, oxidation to aldehyde, and subsequent formaldehyde condensation followed by reduction to install the hydroxymethyl group at the 4'-position.
- Execute bis-triflation, base-catalyzed cyclization to form the 7-membered ring, chlorination, and final deprotection and acylation to yield the target cytosine analog.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, the adoption of this novel synthetic route offers substantial strategic advantages that directly impact the bottom line and operational resilience. The elimination of chromatographic purification steps is perhaps the most significant cost driver, as it removes the need for expensive silica gel, large volumes of organic solvents, and specialized equipment, thereby drastically reducing the variable costs associated with manufacturing. This simplification of the process also shortens the cycle time for each batch, allowing for faster throughput and increased production capacity without the need for additional capital investment in infrastructure.
- Cost Reduction in Manufacturing: The streamlined nature of this process leads to significant cost savings by minimizing reagent consumption and waste generation. By avoiding chromatography, the process reduces the volume of solvent waste that requires treatment and disposal, aligning with green chemistry principles and reducing environmental compliance costs. The high yields obtained at each step, particularly in the final crystallization stages, ensure that raw material utilization is maximized, further driving down the cost per kilogram of the final API. These efficiencies make the production of complex nucleoside analogs economically viable even in competitive markets.
- Enhanced Supply Chain Reliability: The robustness of this synthetic route enhances supply chain reliability by reducing the risk of batch failures and production delays. The ability to produce intermediates that are stable and easily purified via crystallization ensures a consistent supply of high-quality material, mitigating the risks associated with supply disruptions. Furthermore, the use of commercially available and relatively inexpensive reagents reduces dependency on specialized or scarce raw materials, safeguarding the supply chain against market volatility and geopolitical uncertainties that often affect the availability of exotic chemicals.
- Scalability and Environmental Compliance: The process is inherently scalable, as demonstrated by the successful transition from gram to kilogram scales without loss of efficiency or purity. This scalability is crucial for meeting the growing global demand for antiviral medications. Additionally, the reduced solvent usage and waste generation contribute to a lower environmental footprint, helping pharmaceutical companies meet their sustainability goals and comply with increasingly stringent environmental regulations. The improved bulk density of the final product also reduces storage and transportation costs, adding another layer of logistical efficiency to the supply chain.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis and application of the disclosed nucleoside analogs. These answers are derived directly from the technical specifications and experimental data provided in the patent documentation, ensuring accuracy and relevance for industry professionals. Understanding these aspects is crucial for evaluating the feasibility of integrating this technology into existing manufacturing portfolios.
Q: How does this new process improve impurity profiles compared to conventional methods?
A: The disclosed method significantly reduces impurity levels, with specific impurities reduced by factors of 1.5 to 10 times. Notably, several key impurities present in small-scale syntheses are not detected in the large-scale process, achieving final purity levels up to 99.9%.
Q: Is chromatography required for purifying intermediates in this synthesis route?
A: No, one of the primary advantages of this process is the ability to obtain compound (I) without the use of chromatography for intermediate purification. The method relies on crystallization and extraction, which enhances scalability and reduces production costs.
Q: What are the physical property improvements of the final product?
A: The compound produced via this method exhibits improved crystallinity, with an increase of 20 to 60% compared to small-scale preparations. Additionally, the bulk density is enhanced, which improves the flowability of the material during downstream formulation processing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Substituted Nucleoside Analogs Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of having a reliable partner who can translate complex patent technologies into commercial reality. Our team of expert process chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project moves seamlessly from the laboratory to the manufacturing plant. We are committed to delivering high-purity intermediates and APIs that meet stringent purity specifications, supported by our rigorous QC labs and state-of-the-art analytical capabilities. Our facility is equipped to handle the specific reagents and conditions required for nucleoside synthesis, including low-temperature reactions and sensitive halogenation steps.
We invite you to collaborate with us to optimize your supply chain and reduce your manufacturing costs. By leveraging our expertise in process development and scale-up, we can help you implement this advanced synthetic route to achieve superior product quality and operational efficiency. Please contact our technical procurement team to request a Customized Cost-Saving Analysis tailored to your specific needs. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our capabilities align with your project requirements and timelines.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →