Scalable Synthesis of Chiral SHP2 Inhibitor Intermediates via Asymmetric Methylation
Scalable Synthesis of Chiral SHP2 Inhibitor Intermediates via Asymmetric Methylation
The pharmaceutical industry is constantly seeking robust and scalable pathways for the production of complex chiral scaffolds, particularly those serving as key building blocks for next-generation oncology therapeutics. Patent CN113717178A, published on November 30, 2021, introduces a significant advancement in the preparation of intermediates for SHP2 (Src Homology 2 domain-containing phosphatase-2) inhibitors. This intellectual property outlines a novel synthetic strategy that leverages asymmetric induction to construct the sterically demanding 7-azabicyclo[2.2.1]heptane ring system with high stereochemical fidelity. By utilizing tert-butylsulfinamide as a chiral auxiliary, the disclosed method circumvents the traditional pitfalls of racemic synthesis followed by resolution, thereby offering a more direct and atom-economical route to high-value pharmaceutical intermediates. For R&D directors and process chemists, this patent represents a critical methodological reference for accessing enantiopure amines that are otherwise challenging to synthesize with consistent quality.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of bridged bicyclic amines like the 7-azabicyclo[2.2.1]heptane core has relied heavily on the resolution of racemic mixtures, a process that inherently caps the maximum theoretical yield at 50% and generates substantial chemical waste. Conventional alkylation strategies often suffer from poor diastereocontrol, leading to complex mixtures of isomers that require expensive and time-consuming purification techniques such as preparative chiral HPLC or repeated recrystallization. Furthermore, traditional routes may involve harsh reaction conditions or unstable intermediates that pose safety risks during scale-up, complicating the supply chain for clinical trial materials. The reliance on late-stage resolution not only inflates the cost of goods sold (COGS) but also introduces variability in the impurity profile, which is a major concern for regulatory compliance in GMP manufacturing environments.
The Novel Approach
In stark contrast, the methodology described in CN113717178A employs a chiral pool strategy starting from a sulfinylimine precursor, which directs the stereochemical outcome of the subsequent carbon-carbon bond-forming reactions.  This approach allows for the precise installation of the methyl group at the quaternary center with excellent diastereoselectivity, effectively setting the stereochemistry early in the synthesis. The route is characterized by its operational simplicity, utilizing common reagents such as methyl magnesium bromide and di-tert-butyl dicarbonate under mild thermal conditions ranging from 0°C to 60°C. By avoiding the need for chiral chromatography and minimizing the number of unit operations, this novel pathway significantly streamlines the manufacturing process, making it highly attractive for the cost reduction in pharmaceutical intermediate manufacturing required by modern drug development pipelines.
This approach allows for the precise installation of the methyl group at the quaternary center with excellent diastereoselectivity, effectively setting the stereochemistry early in the synthesis. The route is characterized by its operational simplicity, utilizing common reagents such as methyl magnesium bromide and di-tert-butyl dicarbonate under mild thermal conditions ranging from 0°C to 60°C. By avoiding the need for chiral chromatography and minimizing the number of unit operations, this novel pathway significantly streamlines the manufacturing process, making it highly attractive for the cost reduction in pharmaceutical intermediate manufacturing required by modern drug development pipelines.
Mechanistic Insights into Asymmetric Methylation and Deprotection
The cornerstone of this synthetic sequence is the diastereoselective addition of a methyl nucleophile to the N-tert-butanesulfinyl imine (Compound I). The bulky tert-butyl group on the sulfur atom creates a steric environment that shields one face of the imine double bond, forcing the incoming Grignard reagent to attack from the less hindered trajectory. 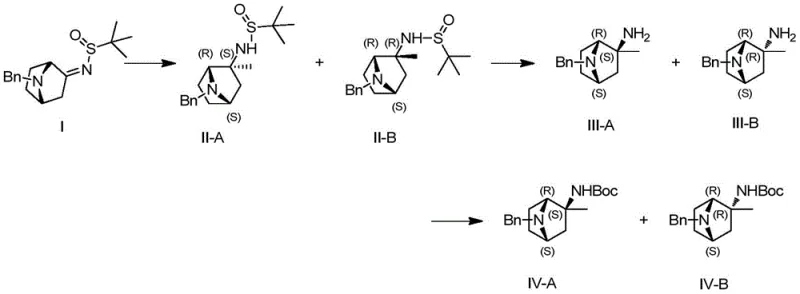 This mechanistic feature ensures that the resulting amine (Compound II) is formed predominantly as a single diastereomer, preserving the chirality throughout the subsequent transformations. Following the methylation, the chiral auxiliary is cleaved under acidic conditions, typically using hydrochloric acid in dioxane, which protonates the nitrogen and facilitates the departure of the sulfinyl group without racemizing the newly formed stereocenter. The resulting free amine (Compound III) is then immediately protected as a Boc-carbamate to prevent side reactions and to enhance the stability of the intermediate for storage and transport.
This mechanistic feature ensures that the resulting amine (Compound II) is formed predominantly as a single diastereomer, preserving the chirality throughout the subsequent transformations. Following the methylation, the chiral auxiliary is cleaved under acidic conditions, typically using hydrochloric acid in dioxane, which protonates the nitrogen and facilitates the departure of the sulfinyl group without racemizing the newly formed stereocenter. The resulting free amine (Compound III) is then immediately protected as a Boc-carbamate to prevent side reactions and to enhance the stability of the intermediate for storage and transport.
Impurity control is intrinsically built into this mechanism due to the high selectivity of the initial addition step, which minimizes the formation of diastereomeric byproducts that are difficult to separate. The subsequent debenzylation step, catalyzed by palladium hydroxide or Pd/C in the presence of a hydrogen donor like ammonium formate or acetic acid, proceeds cleanly to remove the benzyl protecting group from the bridgehead nitrogen. This hydrogenolysis is critical for unveiling the secondary amine functionality required for the final coupling with the SHP2 inhibitor pharmacophore. The final salification with oxalic acid not only isolates the product as a stable crystalline salt (Compound VI) but also serves as a final purification step, as salts often exhibit different solubility profiles that allow for the rejection of trace organic impurities, ensuring the high-purity pharmaceutical intermediate specifications demanded by downstream API synthesis.
How to Synthesize 7-Azabicyclo[2.2.1]heptane Derivatives Efficiently
The synthesis of these valuable chiral intermediates follows a logical five-step progression that balances chemical efficiency with operational safety. The process begins with the activation of the imine followed by nucleophilic attack, proceeds through deprotection and re-protection to manage reactivity, and concludes with hydrogenolysis and salt formation. Each step has been optimized in the patent examples to demonstrate robustness across different scales, from small laboratory flasks to larger reaction vessels. The detailed standardized synthesis steps see the guide below for specific molar ratios and solvent systems that ensure reproducibility.
- React compound I with a methylating agent like methyl magnesium bromide to obtain diastereoisomer II.
- Remove the tert-butyl sulfinyl group from compound II under acidic conditions to yield compound III.
- Protect the amine in compound III with Boc2O under alkaline conditions to form compound IV.
- Perform catalytic debenzylation on compound IV using a palladium catalyst and hydrogen donor to obtain compound V.
- Salify compound V with oxalic acid in an organic solvent to isolate the final intermediate VI.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of the route described in CN113717178A offers tangible benefits regarding cost stability and material availability. The shift from resolution-based methods to asymmetric synthesis fundamentally alters the economics of production by doubling the theoretical yield of the desired enantiomer and eliminating the disposal costs associated with the unwanted isomer. This efficiency translates directly into a more competitive pricing structure for the final active pharmaceutical ingredient, allowing healthcare providers better access to life-saving medications.
- Cost Reduction in Manufacturing: The elimination of chiral separation columns and the reduction in solvent usage per kilogram of product significantly lower the variable costs associated with production. By utilizing inexpensive and widely available reagents such as methyl magnesium bromide and oxalic acid, the process avoids the supply bottlenecks often associated with exotic chiral catalysts. Furthermore, the ability to isolate intermediates as stable salts reduces the risk of material degradation during storage, minimizing write-offs and ensuring that capital is not tied up in spoiled inventory.
- Enhanced Supply Chain Reliability: The synthetic route relies on commodity chemicals and standard unit operations like extraction, distillation, and filtration, which can be easily replicated across multiple manufacturing sites globally. This flexibility mitigates the risk of supply disruption caused by geopolitical issues or single-source dependencies on specialized reagents. The robustness of the chemistry, demonstrated by its tolerance to slight variations in temperature and stoichiometry, ensures consistent batch-to-batch quality, which is essential for maintaining uninterrupted clinical supply chains.
- Scalability and Environmental Compliance: The process operates at moderate temperatures and pressures, reducing the energy footprint of the manufacturing facility and aligning with green chemistry principles. The avoidance of heavy metal catalysts in the early stages and the use of recyclable solvents like ethyl acetate and heptane simplify waste treatment protocols. This environmental compatibility facilitates faster regulatory approvals and supports the sustainability goals of modern pharmaceutical companies, making the commercial scale-up of complex chiral amines both economically and ecologically viable.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthetic technology. These answers are derived directly from the experimental data and claims presented in the patent documentation to provide clarity on the feasibility and scope of the method.
Q: What represents the primary technical advantage of the synthesis route in CN113717178A?
A: The primary advantage is the use of tert-butylsulfinamide chemistry to achieve high diastereoselectivity during the methylation step, eliminating the need for difficult chiral separations of racemic mixtures and ensuring high optical purity of the 7-azabicyclo[2.2.1]heptane core.
Q: Is this synthetic route suitable for large-scale commercial production?
A: Yes, the patent explicitly demonstrates scalability through examples ranging from gram-scale to multi-gram batches using standard reagents like MeMgBr and Pd/C, with reaction conditions maintained at moderate temperatures (0-60°C) that are easily manageable in industrial reactors.
Q: What is the therapeutic application of the final compounds derived from these intermediates?
A: These intermediates are critical precursors for SHP2 allosteric inhibitors, which are being developed as potent anti-tumor agents targeting receptor tyrosine kinase signaling pathways in various cancers.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable SHP2 Inhibitor Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of high-quality intermediates in the development of targeted cancer therapies like SHP2 inhibitors. Our team of expert process chemists has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project transitions smoothly from benchtop discovery to full-scale manufacturing. We maintain stringent purity specifications and operate rigorous QC labs equipped with state-of-the-art analytical instrumentation to guarantee that every batch of 7-azabicyclo[2.2.1]heptane derivative meets the exacting standards required for GMP API synthesis.
We invite you to contact our technical procurement team to discuss how we can support your specific project needs with a Customized Cost-Saving Analysis tailored to your volume requirements. By partnering with us, you gain access to specific COA data and route feasibility assessments that will help you optimize your supply chain and accelerate your time to market. Let us be your strategic partner in delivering the complex building blocks necessary for the next generation of oncology medicines.