Scalable Synthesis of L-Amino Acid Gliotoxin Esters for Next-Generation Oncology Drugs
Scalable Synthesis of L-Amino Acid Gliotoxin Esters for Next-Generation Oncology Drugs
The pharmaceutical landscape is constantly evolving with the discovery of potent natural product derivatives that offer superior therapeutic profiles. A significant breakthrough in this domain is documented in patent CN113087712B, which details the design and synthesis of L-amino acid-6-gliotoxin ester trifluoroacetate compounds. These novel entities are derived from gliotoxin, a secondary metabolite of Aspergillus fumigatus, and represent a strategic advancement in the development of histone lysine demethylase (LSD1) inhibitors. The patent outlines a robust methodology for modifying the gliotoxin scaffold to enhance its antitumor efficacy while mitigating the stability and toxicity issues often associated with epipolythiodiketopiperazines (ETPs). For R&D directors and procurement specialists seeking reliable pharmaceutical intermediate suppliers, understanding the nuances of this synthetic pathway is crucial for securing high-quality inputs for oncology drug pipelines.
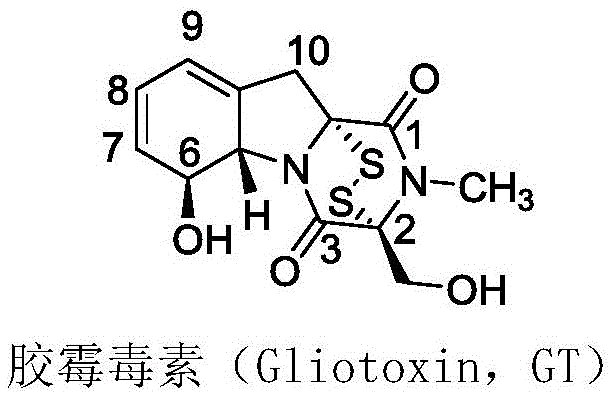
Gliotoxin itself possesses a complex structure characterized by a diketopiperazine skeleton bridged by a disulfide bond, which serves as the key pharmacophore for its biological activity. However, direct clinical application has been hindered by low natural yields and inherent toxicity. The innovation presented in this patent addresses these challenges by introducing amino acid moieties at the 6-position hydroxyl group. This modification not only diversifies the chemical space of ETP compounds but also creates a series of derivatives (General Formula I) that demonstrate markedly improved inhibitory activity against LSD1. This enzyme is a known driver in various aggressive tumors, including esophageal, gastric, lung, and breast cancers, making these derivatives highly valuable candidates for next-generation antitumor therapeutics.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the utilization of gliotoxin and related ETP compounds has been constrained by significant pharmacological and supply chain hurdles. Native gliotoxin, while biologically active, suffers from poor chemical stability due to the reactivity of its disulfide bridge and polysulfide linkages. Furthermore, its clinical translation is limited by considerable toxic side effects and the difficulty in obtaining sufficient quantities through traditional fermentation methods, which often report very low yields. From a manufacturing perspective, relying on unmodified natural extracts introduces variability in purity and supply continuity, posing risks for large-scale API manufacturing. The lack of structural diversity in early-stage research also meant that optimizing the therapeutic index—balancing efficacy with safety—was nearly impossible without a versatile synthetic platform to generate analogues.
The Novel Approach
The methodology described in patent CN113087712B offers a transformative solution by employing a semi-synthetic strategy that leverages optimized fermentation followed by precise chemical modification. By isolating gliotoxin from a high-yield strain of Aspergillus fumigatus ZSS02, the process ensures a steady supply of the core scaffold. The subsequent chemical derivatization allows for the systematic exploration of structure-activity relationships (SAR) through the attachment of various L-amino acids. This approach effectively decouples the production of the complex core from the final diversification step, enabling the creation of a library of compounds with tailored properties. The resulting L-amino acid-6-gliotoxin esters exhibit enhanced stability and reduced toxicity while maintaining or improving the critical LSD1 inhibitory activity, thereby overcoming the primary bottlenecks of the parent compound.
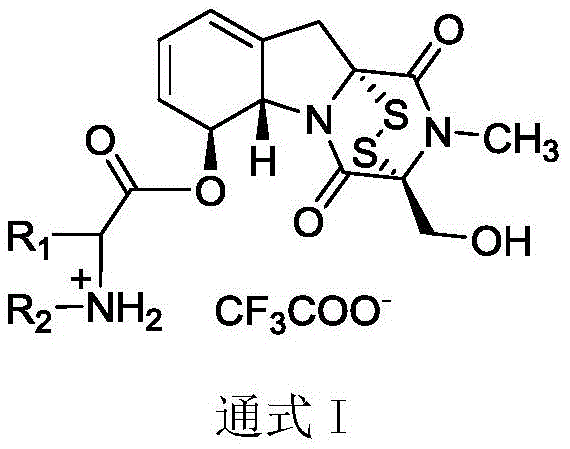
Mechanistic Insights into Chemoselective Esterification
The core of this synthetic innovation lies in the chemoselective esterification of the 6-position hydroxyl group on the gliotoxin molecule. This reaction must be executed with extreme precision to avoid disrupting the sensitive disulfide bond, which is the active center responsible for the compound's biological function. The process utilizes N-Boc-L-amino acids as coupling partners, activated by dicyclohexylcarbodiimide (DCC) and catalyzed by 4-dimethylaminopyridine (DMAP) in dichloromethane. This specific combination of reagents facilitates the formation of the ester linkage under mild conditions, typically at room temperature, which is vital for preserving the integrity of the polysulfide bridge. The use of the Boc (tert-butyloxycarbonyl) protecting group on the amino acid nitrogen ensures that the amine functionality remains inert during the coupling, preventing unwanted side reactions such as amide formation or polymerization.
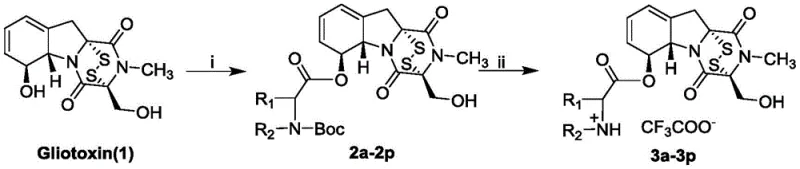
Following the successful formation of the protected intermediate (compounds 2a-2p), the synthesis proceeds to a deprotection step that simultaneously generates the final salt form. Treatment with excess trifluoroacetic acid (TFA) efficiently removes the Boc group, liberating the free amine which immediately forms a stable trifluoroacetate salt. This step is critical not only for revealing the cationic amino acid moiety but also for enhancing the water solubility and pharmaceutical handling properties of the final product. The entire sequence is designed to maximize yield and purity, with column chromatography employed to isolate the intermediates before the final salt formation. This rigorous purification protocol ensures that the final high-purity pharmaceutical intermediates meet the stringent quality standards required for preclinical and clinical evaluation, minimizing the risk of impurity-related toxicity.
How to Synthesize L-Amino Acid-6-Gliotoxin Esters Efficiently
The synthesis of these potent LSD1 inhibitors follows a streamlined two-step protocol that balances efficiency with the preservation of stereochemical integrity. The process begins with the activation of N-Boc-amino acids and their subsequent coupling to the gliotoxin core, followed by a straightforward acidic workup to yield the target trifluoroacetate salts. This route is particularly advantageous for contract development and manufacturing organizations (CDMOs) looking to establish robust supply chains for oncology ingredients. The detailed standardized synthesis steps for producing these compounds are outlined in the guide below, providing a clear roadmap for laboratory and pilot-scale production.
- Dissolve Gliotoxin in dichloromethane and react with N-Boc-L-amino acids using DCC and DMAP catalysts to form the protected ester intermediate.
- Purify the intermediate product (2a-2p) via column chromatography to ensure high chemical purity before the final step.
- Treat the intermediate with excess trifluoroacetic acid in dichloromethane to remove the Boc group and form the final trifluoroacetate salt (3a-3p).
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this synthetic route offers distinct strategic advantages over sourcing native gliotoxin or developing entirely de novo synthetic pathways. The semi-synthetic nature of the process leverages a renewable biological starting material, reducing reliance on complex total synthesis which can be cost-prohibitive and environmentally taxing. The optimization of the fermentation process to achieve yields of 208mg/L significantly lowers the cost basis of the starting material, creating a favorable economic environment for downstream chemical processing. Furthermore, the use of common organic solvents and standard coupling reagents ensures that the manufacturing process can be easily scaled using existing infrastructure without requiring specialized high-pressure or cryogenic equipment.
- Cost Reduction in Manufacturing: The elimination of expensive transition metal catalysts and the use of mild reaction conditions contribute to substantial cost savings. By avoiding harsh conditions that could degrade the sensitive disulfide bridge, the process minimizes waste and rework, leading to higher overall throughput. The ability to use a variety of commercially available N-Boc-amino acids allows for flexible sourcing strategies, mitigating the risk of single-supplier dependency for key reagents. Additionally, the formation of stable trifluoroacetate salts simplifies the isolation and drying processes, reducing energy consumption and processing time compared to free base formulations.
- Enhanced Supply Chain Reliability: The reliance on a fermentation-derived starting material, produced under optimized conditions, ensures a consistent and scalable supply of the gliotoxin core. This biological foundation provides a buffer against the volatility often seen in petrochemical-based supply chains. The modular nature of the synthesis, where different amino acids can be swapped in the first step, allows manufacturers to rapidly pivot production between different derivatives (3a-3p) based on market demand without retooling the entire facility. This flexibility is crucial for maintaining continuity of supply in the fast-paced oncology drug development sector.
- Scalability and Environmental Compliance: The synthetic route is inherently green, utilizing dichloromethane which can be recovered and recycled, and generating minimal hazardous waste compared to heavy metal-catalyzed alternatives. The simplicity of the workup procedures, involving standard aqueous washes and precipitation, facilitates easy scale-up from grams to kilograms. This scalability ensures that the commercial scale-up of complex pharmaceutical intermediates can be achieved with predictable timelines and reduced regulatory hurdles regarding environmental discharge, aligning with modern sustainability goals in fine chemical manufacturing.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of L-amino acid-6-gliotoxin ester trifluoroacetates. These insights are derived directly from the technical specifications and experimental data provided in the patent documentation, ensuring accuracy for R&D and procurement decision-making. Understanding these details helps stakeholders evaluate the feasibility of integrating these intermediates into their drug discovery programs.
Q: How does the new derivative improve upon native Gliotoxin?
A: The L-amino acid-6-gliotoxin ester derivatives exhibit significantly higher inhibitory activity against LSD1 compared to the parent compound, while offering improved chemical stability and reduced toxic side effects, making them more viable for clinical antitumor applications.
Q: What is the critical structural feature preserved during synthesis?
A: The synthesis specifically targets the 6-position hydroxyl group for esterification while strictly preserving the disulfide bond in the active center, which is essential for the biological activity of epipolythiodiketopiperazine compounds.
Q: Is the fermentation yield of the starting material sufficient for scale-up?
A: Yes, the patent describes optimized fermentation conditions for Aspergillus fumigatus ZSS02 that achieve a gliotoxin yield of 208mg/L, which is substantially higher than literature reports, ensuring a robust supply of the starting material.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable L-Amino Acid-6-Gliotoxin Ester Supplier
At NINGBO INNO PHARMCHEM, we recognize the immense potential of the synthetic pathway described in patent CN113087712B for advancing oncology therapeutics. As a premier CDMO partner, we possess the extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from benchtop discovery to clinical supply is seamless. Our facilities are equipped with state-of-the-art fermentation and chemical synthesis units capable of handling sensitive sulfur-containing compounds with the utmost care. We adhere to stringent purity specifications and operate rigorous QC labs to guarantee that every batch of L-amino acid-6-gliotoxin ester meets the highest international standards for pharmaceutical intermediates.
We invite you to collaborate with us to optimize your supply chain for these high-value antitumor agents. Our technical team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and purity needs. By partnering with us, you gain access to specific COA data and comprehensive route feasibility assessments that can accelerate your project timelines. Contact our technical procurement team today to discuss how we can support your development of next-generation LSD1 inhibitors and secure a reliable supply of these critical building blocks.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →