Advanced Synthesis and Commercial Scale-up of Potent Heterocyclic Thrombin Inhibitors
Advanced Synthesis and Commercial Scale-up of Potent Heterocyclic Thrombin Inhibitors
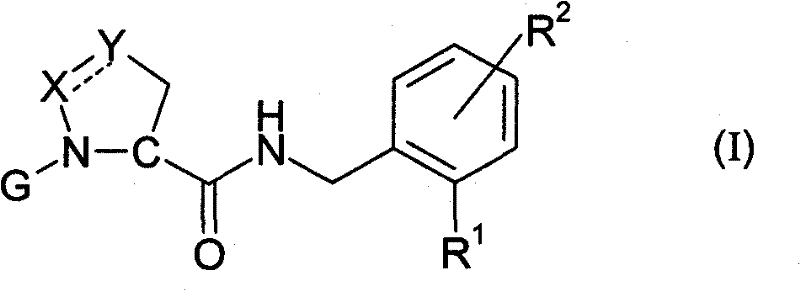
The pharmaceutical landscape for anticoagulant therapy has been significantly reshaped by the discovery of novel heterocyclic carboxamides, as detailed in patent CN102131802A. This intellectual property introduces a sophisticated class of compounds designed as competitive inhibitors of trypsin-like serine proteases, with a specific focus on thrombin. The core innovation lies in the structural integration of a 5-membered heterocyclic ring, such as pyrazoline or isoxazolidine, linked to a functionalized benzylamine moiety containing a tetrazole group. This architecture is not merely a theoretical construct but represents a tangible breakthrough in creating small molecules that mimic the binding properties of larger peptides while offering superior pharmacokinetic profiles. For R&D directors and procurement specialists, understanding the depth of this chemistry is crucial for securing a reliable pipeline of high-purity pharmaceutical intermediates. The patent outlines a robust synthetic methodology that balances complexity with manufacturability, ensuring that these potent inhibitors can be produced consistently to meet stringent regulatory standards.
The development of effective thrombin inhibitors has historically faced significant hurdles, primarily revolving around the limitations of conventional peptidomimetic approaches. Traditional methods often relied on linear peptide sequences that, while potent in vitro, suffered from poor oral bioavailability and rapid metabolic clearance in vivo. These molecules were susceptible to proteolytic degradation, necessitating frequent dosing or parenteral administration, which severely limited their clinical utility and commercial viability. Furthermore, the synthesis of complex peptide chains often involved cumbersome protection and deprotection strategies, leading to low overall yields and high production costs. The accumulation of impurities during these multi-step processes posed additional challenges for purification, often requiring extensive chromatographic separation that is difficult to translate to an industrial scale. Consequently, there was a pressing need for a new chemical paradigm that could retain high affinity for the thrombin active site while overcoming these pharmacokinetic and manufacturing bottlenecks.
In contrast to these legacy approaches, the novel strategy presented in the patent leverages a rigid heterocyclic scaffold to lock the molecule into a bioactive conformation. By replacing flexible peptide bonds with stable amide linkages attached to a cyclic core, the new compounds achieve enhanced metabolic stability and improved membrane permeability. The introduction of the tetrazole ring as a bioisostere for the carboxylic acid group further optimizes the electrostatic interactions within the enzyme's S1 pocket, resulting in nanomolar potency. From a manufacturing perspective, this structural simplification allows for a more convergent synthesis. Instead of building the molecule linearly, key fragments such as the heterocyclic acid and the substituted benzylamine can be prepared independently and coupled in a late stage. This modularity not only streamlines the process but also facilitates the rapid generation of analogues for structure-activity relationship studies, accelerating the drug discovery timeline significantly.
Mechanistic Insights into Amide Coupling and Heterocyclic Cyclization
The chemical mechanism underpinning the synthesis of these inhibitors is a testament to modern organic synthesis efficiency, particularly in the formation of the critical amide bond and the construction of the heterocyclic ring. The formation of the 4,5-dihydro-1H-pyrazole core, for instance, often involves a 1,3-dipolar cycloaddition reaction. As illustrated in the synthesis of Example 1, the reaction of an acrylate derivative with trimethylsilyldiazomethane generates a reactive intermediate that cyclizes to form the pyrazoline ring system. This step is pivotal as it establishes the stereochemistry at the C5 position, which is critical for biological activity. The subsequent acylation of the nitrogen atom on the heterocycle requires precise control to avoid over-acylation or racemization. The use of activated acid chlorides or mixed anhydrides ensures that the acyl group is transferred efficiently to the nucleophilic nitrogen, preserving the integrity of the adjacent chiral centers.
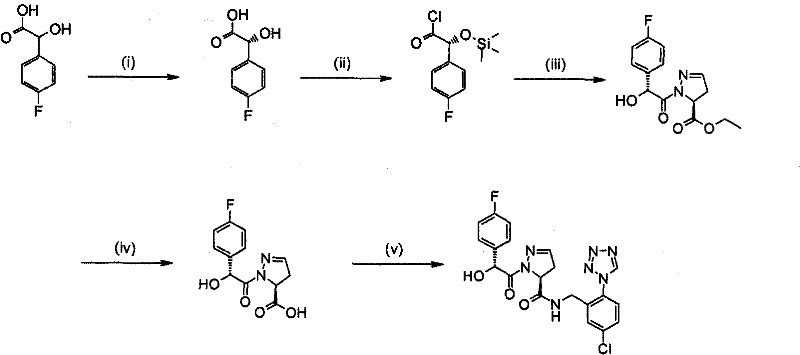
Impurity control is another cornerstone of this mechanistic approach, ensuring that the final API meets rigorous quality specifications. During the amide coupling step, potential side reactions such as epimerization at the alpha-carbon of the acylating agent can occur if the reaction conditions are too basic or prolonged. The patent specifies the use of coupling reagents like TBTU, HATU, or EDC in conjunction with additives like HOBt or HOAt to suppress these pathways. These additives form active esters that react rapidly with the amine, minimizing the lifetime of the activated species and thus reducing the risk of racemization. Furthermore, the purification strategy often relies on selective crystallization rather than solely on chromatography. For example, the formation of ammonium salts or specific solvates can be exploited to precipitate the desired enantiomer while leaving impurities in the mother liquor. This physical separation method is far more scalable and cost-effective for commercial production than preparative HPLC.
How to Synthesize Heterocyclic Thrombin Inhibitors Efficiently
The practical execution of this synthesis requires a deep understanding of reaction kinetics and thermodynamic control to maximize yield and purity. The process typically begins with the preparation of the chiral heterocyclic acid fragment, which serves as the P2 mimic in the inhibitor design. This fragment is then activated and coupled with the P1 benzylamine component, which carries the essential tetrazole pharmacophore. The success of this coupling depends heavily on the choice of solvent and base; polar aprotic solvents like DMF or acetonitrile are often preferred to solubilize the zwitterionic intermediates, while non-nucleophilic bases like DIPEA or NMM are used to scavenge the generated acid without attacking the activated ester. Detailed standardized synthesis steps are provided in the guide below to ensure reproducibility across different manufacturing sites.
- Preparation of the heterocyclic core via cycloaddition using trimethylsilyldiazomethane and subsequent acylation.
- Coupling of the heterocyclic acid with the tetrazole-substituted benzylamine using reagents like TBTU or EDC.
- Purification of the final compound through crystallization or flash chromatography to ensure high purity.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the transition to this novel synthetic route offers substantial strategic benefits beyond mere chemical elegance. The primary advantage lies in the significant reduction of manufacturing complexity, which directly translates to cost optimization. By eliminating the need for expensive transition metal catalysts often required in cross-coupling reactions for other classes of inhibitors, the process relies on readily available organic reagents. This shift reduces the burden of heavy metal testing and removal, streamlining the quality control workflow and shortening the release time for batches. Additionally, the convergent nature of the synthesis means that supply chain risks are mitigated; if one fragment faces a raw material shortage, the other can still be stockpiled, providing flexibility in production planning and inventory management.
Enhanced supply chain reliability is further bolstered by the robustness of the purification protocols described in the patent. Traditional peptide syntheses often result in complex mixtures of deletion sequences and byproducts that are difficult to separate. In contrast, the small molecule nature of these heterocyclic carboxamides allows for sharp melting points and distinct solubility profiles. This physical characteristic enables the use of recrystallization as a primary purification tool, which is inherently more scalable than column chromatography. Large-scale crystallization tanks can process hundreds of kilograms of material in a single batch, ensuring a consistent supply of high-purity intermediates. This scalability is crucial for meeting the demands of global clinical trials and eventual commercial launch without the need for extensive process re-engineering.
Scalability and environmental compliance are also key drivers for adopting this technology. The synthetic route minimizes the use of hazardous solvents where possible and employs reagents that generate manageable waste streams. For instance, the byproducts of carbodiimide-mediated couplings are urea derivatives that are generally non-toxic and easy to remove. This aligns with modern green chemistry principles and reduces the environmental footprint of the manufacturing process. Furthermore, the stability of the final compounds simplifies logistics; unlike sensitive biologics or unstable peptides, these small molecules can be stored under standard conditions without requiring cold chain transportation. This durability reduces shipping costs and expands the geographical reach of the supply network, ensuring that partners worldwide have access to reliable pharmaceutical intermediates.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these thrombin inhibitors. These answers are derived directly from the technical specifications and experimental data provided in the patent documentation. They are intended to clarify the feasibility of large-scale production, the stability of the compounds, and the regulatory considerations for their use in drug development. Understanding these aspects is vital for stakeholders evaluating the potential of this technology for their own pipelines.
Q: What are the key stability advantages of these heterocyclic carboxamides?
A: These compounds exhibit superior metabolic stability compared to traditional peptide mimetics due to the rigid heterocyclic scaffold which resists enzymatic degradation.
Q: Is the synthesis route scalable for commercial production?
A: Yes, the process utilizes standard amide coupling reagents and avoids exotic catalysts, making it highly amenable to multi-kilogram scale-up in GMP facilities.
Q: How is the stereochemical purity controlled during manufacturing?
A: Stereocontrol is achieved through the use of chiral starting materials and specific crystallization steps that enrich the desired enantiomer to over 99% ee.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Heterocyclic Carboxamides Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of translating innovative patent chemistry into commercial reality. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project moves seamlessly from the laboratory bench to the manufacturing floor. We understand that the synthesis of complex heterocyclic systems requires precise control over reaction parameters and purification techniques. Our state-of-the-art facilities are equipped with rigorous QC labs capable of detecting impurities at trace levels, guaranteeing that every batch meets stringent purity specifications. Whether you require custom synthesis of specific analogues or large-scale supply of the core intermediates, our infrastructure is designed to support your long-term goals.
We invite you to initiate a dialogue with our technical procurement team to discuss how we can optimize your supply chain for these valuable anticoagulant intermediates. By partnering with us, you gain access to a Customized Cost-Saving Analysis that identifies specific opportunities to reduce expenses without compromising quality. We encourage you to request specific COA data and route feasibility assessments to validate our capabilities against your internal standards. Let us demonstrate how our expertise in heterocyclic chemistry can accelerate your development timeline and secure a competitive advantage in the cardiovascular therapeutic market.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →