Advanced Purification Technology for 2'-Fluoro-2'-Deoxyuridine: Scaling High-Purity Pharmaceutical Intermediates
Advanced Purification Technology for 2'-Fluoro-2'-Deoxyuridine: Scaling High-Purity Pharmaceutical Intermediates
In the highly competitive landscape of antiviral drug development, the availability of high-purity nucleoside intermediates is a critical bottleneck that often dictates project timelines and commercial viability. Patent CN112409420A introduces a transformative purification methodology specifically designed for 2'-fluoro-2'-deoxyuridine, a key building block in the synthesis of various therapeutic agents. This technical disclosure addresses the longstanding challenge of isolating this compound from crude reaction mixtures, which typically contain stubborn impurities and exhibit poor physical properties for isolation. By leveraging a strategic protection-deprotection sequence, the patented process achieves product purity levels exceeding 98.5 percent, setting a new benchmark for quality in pharmaceutical intermediate manufacturing. For R&D directors and procurement specialists seeking a reliable pharmaceutical intermediate supplier, understanding the mechanistic advantages of this route is essential for securing a stable supply chain.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditionally, the synthesis of 2'-fluoro-2'-deoxyuridine involves the fluorination of uridine, a process that inevitably generates a complex mixture of byproducts and unreacted starting materials. The fundamental issue lies in the physicochemical nature of the crude nucleoside product, which is notoriously sticky, hygroscopic, and resistant to forming stable crystal lattices. When manufacturers attempt to purify this crude oil using standard recrystallization techniques, the material often remains as an amorphous gum or oil, trapping impurities within its matrix and preventing effective separation. Furthermore, traditional chromatographic purification, while effective on a laboratory scale, is economically prohibitive and operationally cumbersome for multi-kilogram or ton-scale production due to high solvent consumption and low throughput. These limitations result in inconsistent batch quality, extended lead times, and significantly elevated manufacturing costs, creating substantial risks for downstream API production.
The Novel Approach
The innovative strategy outlined in the patent circumvents these physical barriers by temporarily modifying the chemical structure of the target molecule to facilitate isolation. Instead of struggling to crystallize the polar, sticky 2'-fluoro-2'-deoxyuridine directly, the process converts it into a less polar, crystalline di-ester derivative, referred to as Compound 2. This structural transformation dramatically improves the solid-state properties of the molecule, allowing it to precipitate cleanly from solution upon cooling. 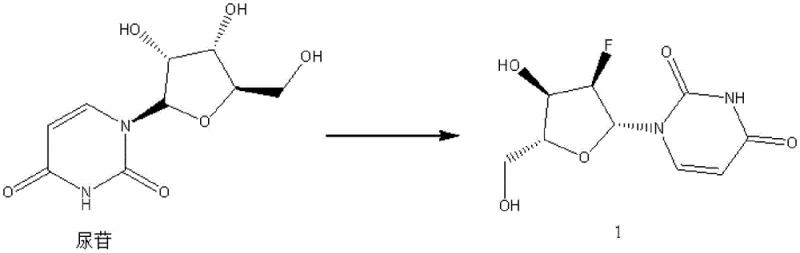 Once the intermediate is isolated in high purity via simple filtration and washing, the protecting groups are removed under mild hydrolytic conditions to regenerate the parent nucleoside. This approach effectively decouples the purification challenge from the final product's difficult physical properties, enabling a robust, scalable, and cost-effective manufacturing workflow that delivers consistent high-purity material suitable for stringent pharmaceutical applications.
Once the intermediate is isolated in high purity via simple filtration and washing, the protecting groups are removed under mild hydrolytic conditions to regenerate the parent nucleoside. This approach effectively decouples the purification challenge from the final product's difficult physical properties, enabling a robust, scalable, and cost-effective manufacturing workflow that delivers consistent high-purity material suitable for stringent pharmaceutical applications.
Mechanistic Insights into Esterification-Protection and Hydrolysis
The core of this purification technology relies on the regioselective esterification of the hydroxyl groups at the 3' and 4' positions of the ribose sugar moiety. By reacting the crude 2'-fluoro-2'-deoxyuridine with an acylating agent such as acetic anhydride or benzoyl chloride in the presence of a base like triethylamine, the polar hydroxyl groups are masked as esters. This reduction in polarity not only enhances the solubility profile in organic solvents like ethyl acetate but also promotes intermolecular interactions that favor crystal lattice formation. The use of catalysts such as 4-dimethylaminopyridine (DMAP) further accelerates the acylation kinetics, ensuring complete conversion even at controlled low temperatures between 0 and 20°C, which minimizes the risk of side reactions or degradation of the sensitive glycosidic bond. 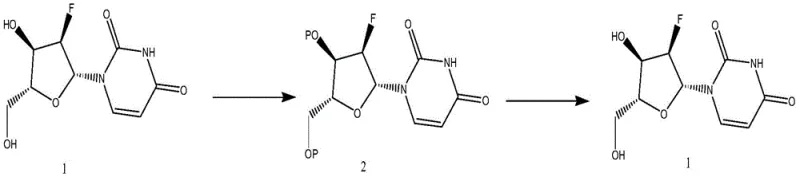 The resulting intermediate, Compound 2, possesses distinct crystallization characteristics that allow impurities to remain in the mother liquor during the cooling phase, typically conducted between -20 and 10°C.
The resulting intermediate, Compound 2, possesses distinct crystallization characteristics that allow impurities to remain in the mother liquor during the cooling phase, typically conducted between -20 and 10°C.
Following the isolation of the purified intermediate, the final step involves a saponification reaction to restore the original hydroxyl functionality. Dissolving the crystalline Compound 2 in methanol and treating it with sodium hydroxide facilitates a rapid and clean hydrolysis of the ester bonds. This step is critical because it must proceed quantitatively without affecting the fluorine atom at the 2' position or the uracil base. The patent data indicates that this hydrolysis proceeds with exceptional efficiency, yielding the final 2'-fluoro-2'-deoxyuridine with recoveries often exceeding 93 percent and purity levels consistently above 98.5 percent. The simplicity of the workup, involving merely filtration and drying after hydrolysis, underscores the elegance of this chemical design, offering a practical solution for removing trace impurities that would otherwise persist through conventional processing methods.
How to Synthesize 2'-Fluoro-2'-Deoxyuridine Efficiently
Implementing this purification protocol requires precise control over reaction stoichiometry and temperature profiles to maximize the yield of the crystalline intermediate. The process begins by dissolving the crude nucleoside in ethyl acetate, followed by the addition of triethylamine to scavenge the acid byproduct generated during acylation. The acylating reagent is added dropwise to manage the exotherm and ensure uniform reaction progress. Once the protected intermediate is formed and crystallized, it is subjected to basic hydrolysis in methanol. This standardized approach ensures that the transition from crude oil to high-purity solid is reproducible across different batch sizes, providing a clear pathway for process chemists to adopt this technology in their own facilities.
- Dissolve crude 2'-fluoro-2'-deoxyuridine in ethyl acetate, add triethylamine, and dropwise add an acylating reagent (acetic anhydride or benzoyl chloride) at 0-20°C to form the protected intermediate.
- Add methanol to the reaction mixture, stir, and cool to -20 to 10°C to induce crystallization of the intermediate compound, then filter and wash with ethyl acetate.
- Dissolve the purified intermediate in methanol, add sodium hydroxide to hydrolyze the protecting groups, filter the final product, and dry to obtain high-purity 2'-fluoro-2'-deoxyuridine.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this purification method translates into tangible operational efficiencies and risk mitigation strategies. The primary advantage lies in the elimination of complex and costly purification steps such as preparative HPLC or extensive column chromatography, which are often required for sticky nucleoside intermediates. By shifting the purification burden to a crystallizable intermediate, the process utilizes standard solid-liquid separation equipment, drastically reducing solvent usage and processing time. This simplification of the unit operations directly correlates to a significant reduction in manufacturing costs, as the reliance on expensive consumables and specialized labor is minimized. Furthermore, the high recovery rates observed in the hydrolysis step ensure that raw material utilization is optimized, contributing to a more sustainable and economically viable production model.
- Cost Reduction in Manufacturing: The transition to a crystallization-based purification strategy eliminates the need for expensive chromatographic resins and large volumes of high-grade solvents typically associated with purifying polar nucleosides. By utilizing commodity chemicals like ethyl acetate, methanol, and acetic anhydride, the overall cost of goods sold (COGS) is substantially lowered. The ability to filter and wash the intermediate solid allows for the efficient removal of impurities without the yield losses inherent in multiple chromatographic passes, ensuring that the final cost per kilogram of the API intermediate is highly competitive in the global market.
- Enhanced Supply Chain Reliability: The robustness of this chemical process ensures consistent batch-to-batch quality, which is paramount for maintaining uninterrupted API production schedules. Because the method relies on well-understood chemical transformations and standard equipment, the risk of batch failure due to equipment malfunction or operator error is significantly reduced. This reliability allows supply chain planners to forecast inventory levels with greater confidence, reducing the need for excessive safety stock and minimizing the risk of stockouts that could delay downstream drug development programs or commercial launches.
- Scalability and Environmental Compliance: From an environmental and scalability perspective, this method offers distinct advantages by reducing the generation of hazardous waste streams. The solvents used, primarily ethyl acetate and methanol, are readily recoverable and recyclable, aligning with green chemistry principles and reducing the environmental footprint of the manufacturing process. The straightforward nature of the reaction steps facilitates seamless scale-up from pilot plant to commercial production scales, ensuring that supply can be rapidly expanded to meet market demand without the need for complex process re-engineering or specialized infrastructure investments.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the purification of 2'-fluoro-2'-deoxyuridine, based on the detailed specifications provided in the patent literature. Understanding these nuances helps stakeholders evaluate the feasibility of integrating this intermediate into their specific synthetic routes. These insights are derived directly from the experimental data and process descriptions to ensure accuracy and relevance for technical decision-makers.
Q: Why is direct crystallization of 2'-fluoro-2'-deoxyuridine difficult?
A: Direct crystallization is challenging because nucleoside compounds like 2'-fluoro-2'-deoxyuridine are extremely viscous and tend to form oils rather than solids. Additionally, reaction impurities often co-precipitate, making it nearly impossible to achieve high purity without a derivatization step.
Q: What is the role of the esterification step in this purification method?
A: The esterification step introduces protecting groups (such as acetyl or benzoyl) at the 3' and 4' positions. This chemical modification significantly alters the physical properties of the molecule, transforming the sticky crude oil into a solid, crystalline intermediate that can be easily filtered and washed to remove impurities.
Q: Can this purification method be scaled for industrial production?
A: Yes, the method is highly suitable for industrial scale-up. It utilizes common solvents like ethyl acetate and methanol, avoids complex chromatography, and relies on standard unit operations such as filtration and crystallization, ensuring robust reproducibility and cost-efficiency at large volumes.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2'-Fluoro-2'-Deoxyuridine Supplier
At NINGBO INNO PHARMCHEM, we recognize that the quality of your final therapeutic product is only as good as the intermediates you start with. Our team of expert process chemists has extensively analyzed advanced purification technologies, including the esterification-hydrolysis route described in patent CN112409420A, to ensure we deliver the highest standards of purity and consistency. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, leveraging our state-of-the-art facilities and rigorous QC labs to meet stringent purity specifications. Our commitment to technical excellence means that every batch of 2'-fluoro-2'-deoxyuridine we supply undergoes comprehensive analytical testing to guarantee it meets the demanding requirements of modern antiviral drug synthesis.
We invite pharmaceutical partners to collaborate with us to optimize their supply chains and reduce overall manufacturing costs. By engaging with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. We encourage you to reach out today to discuss your project needs,索取 specific COA data, and review our detailed route feasibility assessments. Let us be your strategic partner in bringing high-quality nucleoside therapeutics to market faster and more efficiently.