Advanced Synthesis of Flavonoid Intermediates for Commercial Scale-up
The escalating challenge of multidrug resistance (MDR) in oncology has necessitated the development of sophisticated chemosensitizers capable of reversing P-glycoprotein-mediated efflux mechanisms. Among the most promising candidates are flavonoid dimer compounds, specifically those linked by poly(ethylene glycol) spacers, which have demonstrated potent inhibitory effects against resistant leukemia and breast cancer cell lines. However, the clinical translation of these molecules has been historically stifled by inefficient synthetic methodologies that fail to meet the rigorous demands of pharmaceutical manufacturing. Patent CN101570529B addresses this critical bottleneck by disclosing a novel, high-efficiency preparation method for key flavonoid intermediates, specifically the 7-benzyl-5,4'-dihydroxyapigenin derivative (Formula B). This technological breakthrough not only simplifies the synthetic architecture but also fundamentally alters the economic feasibility of producing these high-value pharmaceutical intermediates. By leveraging a unique reverse-addition hydrolysis technique, the patent enables the simultaneous removal of specific ester protecting groups that were previously considered kinetically inert under standard conditions. For R&D directors and procurement strategists alike, this represents a pivotal shift from laboratory curiosity to viable commercial asset, offering a pathway to secure the supply chains necessary for next-generation anticancer therapies.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior to the innovations detailed in CN101570529B, the synthesis of flavonoid dimers was plagued by prohibitive complexity and dismal overall yields. As illustrated in the legacy synthetic pathways, the preparation of the target dimer (Formula D, where n=4) typically required a tedious 7-step sequence that resulted in a cumulative yield of merely 9.7%. 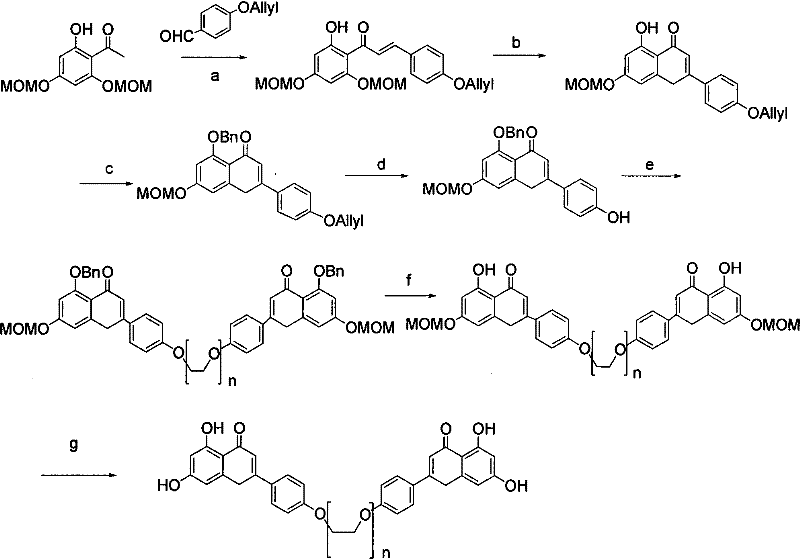 . These conventional routes often relied on the use of expensive starting materials and introduced hazardous reagents, such as iodine-containing compounds, which posed significant challenges for downstream purification and clinical safety. The presence of residual iodine is particularly detrimental in pharmaceutical applications, as it can lead to toxic side effects and necessitates costly additional purification steps to meet regulatory standards. Furthermore, the reliance on multiple protection and deprotection cycles increased the operational burden, generating substantial chemical waste and extending production lead times. For a reliable pharmaceutical intermediate supplier, managing such a fragile and low-yielding process presents an unacceptable risk to supply continuity and cost stability, rendering the final active pharmaceutical ingredient (API) economically unviable for widespread clinical use.
. These conventional routes often relied on the use of expensive starting materials and introduced hazardous reagents, such as iodine-containing compounds, which posed significant challenges for downstream purification and clinical safety. The presence of residual iodine is particularly detrimental in pharmaceutical applications, as it can lead to toxic side effects and necessitates costly additional purification steps to meet regulatory standards. Furthermore, the reliance on multiple protection and deprotection cycles increased the operational burden, generating substantial chemical waste and extending production lead times. For a reliable pharmaceutical intermediate supplier, managing such a fragile and low-yielding process presents an unacceptable risk to supply continuity and cost stability, rendering the final active pharmaceutical ingredient (API) economically unviable for widespread clinical use.
The Novel Approach
In stark contrast to the cumbersome legacy methods, the novel approach disclosed in the patent streamlines the synthesis into a robust 5-step sequence that achieves a total yield of approximately 30%, representing a more than threefold improvement in efficiency. The cornerstone of this methodology is the strategic design of intermediate compounds (Formula E and Formula F) utilizing hexanoyl or benzoyl protecting groups, which offer superior solubility and reactivity profiles compared to traditional acyl groups. 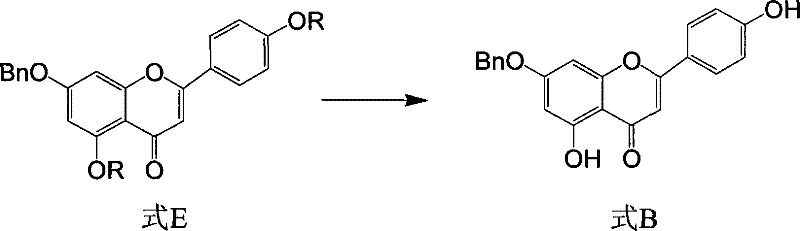 . The critical innovation lies in the conversion of Formula E to Formula B, where a solution of the tri-acylated intermediate is滴加 (dropwise added) into a large excess of sodium alkoxide solution. This specific operational parameter disrupts the thermodynamic equilibrium that typically hinders the hydrolysis of the 5-position ester. By avoiding the introduction of iodine and utilizing commercially abundant reagents like apigenin and benzyl chloride, this route drastically reduces raw material costs and environmental impact. For procurement managers, this translates to a significant cost reduction in API manufacturing, as the process eliminates the need for exotic catalysts and minimizes solvent consumption through higher convergence. The simplicity of the workup procedure, involving standard neutralization and crystallization, further enhances the scalability, making it an ideal candidate for commercial scale-up of complex pharmaceutical intermediates.
. The critical innovation lies in the conversion of Formula E to Formula B, where a solution of the tri-acylated intermediate is滴加 (dropwise added) into a large excess of sodium alkoxide solution. This specific operational parameter disrupts the thermodynamic equilibrium that typically hinders the hydrolysis of the 5-position ester. By avoiding the introduction of iodine and utilizing commercially abundant reagents like apigenin and benzyl chloride, this route drastically reduces raw material costs and environmental impact. For procurement managers, this translates to a significant cost reduction in API manufacturing, as the process eliminates the need for exotic catalysts and minimizes solvent consumption through higher convergence. The simplicity of the workup procedure, involving standard neutralization and crystallization, further enhances the scalability, making it an ideal candidate for commercial scale-up of complex pharmaceutical intermediates.
Mechanistic Insights into Selective Ester Hydrolysis
The success of this synthetic route hinges on a profound understanding of the electronic and steric factors governing flavonoid chemistry, specifically the differential reactivity of phenolic esters. In standard alkaline hydrolysis conditions, the ester groups at the 7 and 4' positions of the flavonoid skeleton are readily cleaved due to their higher acidity and accessibility. However, the ester at the 5-position is notoriously resistant to hydrolysis because the adjacent phenolic hydroxyl group forms a strong intramolecular hydrogen bond with the carbonyl oxygen of the 4-position ketone. This interaction stabilizes the enol form and effectively locks the 5-ester in a conformation that shields it from nucleophilic attack by hydroxide ions. 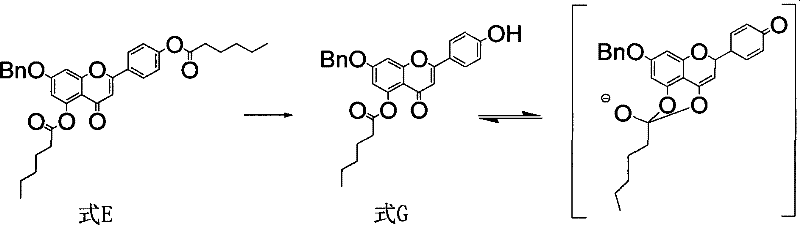 . Consequently, conventional methods often stall at the mono-deprotected stage (Formula G), failing to yield the desired di-hydroxy product (Formula B). The patent elucidates that by employing a reverse addition technique—where the substrate is introduced into a vast excess of base—the local concentration of alkoxide ions is kept sufficiently high to overcome the kinetic barrier imposed by the hydrogen bonding. This forces the simultaneous cleavage of both the 4' and 5-esters before the enol interconversion can sequester the 5-position. This mechanistic nuance is critical for R&D teams aiming to replicate the process, as deviating from the specified addition order or base concentration could result in incomplete reaction and difficult-to-separate impurities. Understanding this kinetic control allows for precise optimization of reaction parameters, ensuring consistent batch-to-batch quality and high purity specifications essential for regulatory filing.
. Consequently, conventional methods often stall at the mono-deprotected stage (Formula G), failing to yield the desired di-hydroxy product (Formula B). The patent elucidates that by employing a reverse addition technique—where the substrate is introduced into a vast excess of base—the local concentration of alkoxide ions is kept sufficiently high to overcome the kinetic barrier imposed by the hydrogen bonding. This forces the simultaneous cleavage of both the 4' and 5-esters before the enol interconversion can sequester the 5-position. This mechanistic nuance is critical for R&D teams aiming to replicate the process, as deviating from the specified addition order or base concentration could result in incomplete reaction and difficult-to-separate impurities. Understanding this kinetic control allows for precise optimization of reaction parameters, ensuring consistent batch-to-batch quality and high purity specifications essential for regulatory filing.
Furthermore, the choice of the acyl protecting group plays a pivotal role in the overall efficiency of the process. The patent highlights that hexanoyl groups (R = -CO(CH2)4CH3) provide an optimal balance of solubility and lability. Unlike shorter acetyl chains which may lead to poor solubility in organic solvents, or bulky benzoyl groups which might require harsher conditions for removal, the hexanoyl moiety ensures that the intermediate (Formula F) remains in solution throughout the benzylation step. This solubility enhancement is not merely a convenience; it is a prerequisite for homogeneous reaction kinetics, which directly correlates to higher yields and fewer side reactions. For process chemists, this means that the reaction can be run at higher concentrations, reducing the volume of solvent required per kilogram of product. This reduction in solvent load has a cascading positive effect on the environmental footprint of the manufacturing process, aligning with modern green chemistry principles. The ability to selectively install a benzyl group at the 7-position while maintaining ester protection at the 5 and 4' positions demonstrates a high level of chemoselectivity, which is often the differentiator between a viable industrial process and a laboratory artifact. This level of control minimizes the formation of regioisomers, thereby simplifying the purification train and reducing the loss of valuable material during chromatography or recrystallization steps.
How to Synthesize 7-Benzyl-5,4'-dihydroxyapigenin Efficiently
The synthesis of the core intermediate Formula B is achieved through a carefully orchestrated sequence that prioritizes solubility management and kinetic control. The process begins with the per-acylation of commercially available apigenin to improve its handling characteristics, followed by a selective benzylation at the 7-position. The final and most critical step involves the simultaneous deprotection of the 5 and 4' esters using the reverse addition hydrolysis method described previously. This approach ensures that the sensitive flavonoid core is preserved while efficiently unveiling the necessary phenolic functionalities for subsequent dimerization. The detailed standardized synthesis steps are provided in the guide below.
- Prepare a solution of the tri-acylated intermediate (Formula E) in an aprotic solvent such as dichloromethane.
- Dropwise add this solution into a large excess of sodium alkoxide alcohol solution (e.g., sodium methoxide in methanol) at 10-40°C.
- Stir for 4-10 hours until TLC confirms consumption, then neutralize and purify to obtain the target phenolic compound.
Commercial Advantages for Procurement and Supply Chain Teams
The implementation of the synthetic route described in CN101570529B offers transformative benefits for the supply chain economics of flavonoid-based therapeutics. By shifting from a 7-step, low-yield process to a streamlined 5-step protocol, manufacturers can realize substantial reductions in production costs without compromising on quality. The elimination of iodine-based reagents removes a significant source of toxicity risk and purification cost, while the use of commodity chemicals like hexanoyl chloride and benzyl chloride ensures that raw material sourcing remains stable and predictable. For supply chain heads, this reliability is paramount, as it mitigates the risk of production stoppages due to the scarcity of specialized reagents. Moreover, the robustness of the reaction conditions, which operate at moderate temperatures (10-40°C) and utilize common solvents like dichloromethane and methanol, facilitates easy technology transfer across different manufacturing sites. This flexibility enhances supply security, allowing for multi-vendor strategies that protect against geopolitical or logistical disruptions. The high overall yield of 30% compared to the historical 9.7% means that less starting material is required to produce the same amount of final API, directly lowering the cost of goods sold (COGS) and improving margin potential for downstream drug developers.
- Cost Reduction in Manufacturing: The dramatic increase in total yield from 9.7% to approximately 30% fundamentally alters the cost structure of the final product. This efficiency gain means that for every kilogram of target dimer produced, the consumption of raw materials and solvents is reduced by nearly two-thirds compared to legacy methods. Additionally, the avoidance of expensive and hazardous iodine reagents eliminates the need for specialized waste treatment and extensive metal scavenging processes, further driving down operational expenditures. The simplified purification workflow, relying on crystallization rather than complex chromatography for intermediates, reduces labor hours and equipment occupancy time. These cumulative savings allow for a more competitive pricing strategy in the global market for oncology intermediates, making potential therapies more accessible while maintaining healthy profit margins for the manufacturer.
- Enhanced Supply Chain Reliability: A major vulnerability in the previous synthetic routes was the dependence on difficult-to-source starting materials and reagents that were prone to supply volatility. The new method utilizes apigenin, a naturally abundant flavonoid, and standard acylating agents that are produced at massive scales for various industries. This shift to commodity-grade inputs ensures that the supply chain is resilient against shortages. Furthermore, the process does not require cryogenic conditions or high-pressure equipment, meaning it can be executed in standard multipurpose chemical plants without the need for capital-intensive infrastructure upgrades. This ease of execution reduces the lead time for scaling up production from pilot plant to commercial tonnage, ensuring that clinical trial material and commercial stock can be delivered on schedule. For procurement managers, this translates to a dependable partner capable of meeting Just-In-Time delivery requirements without the risk of batch failures.
- Scalability and Environmental Compliance: The environmental profile of this synthesis is significantly improved, aligning with increasingly stringent global regulations on chemical manufacturing. By reducing the number of synthetic steps and eliminating heavy metal contaminants, the process generates less hazardous waste and lowers the E-factor (mass of waste per mass of product). The solvents used are common and easily recoverable through distillation, supporting a circular economy approach within the plant. The high solubility of the intermediates allows for reactions to be run at higher concentrations, minimizing the volume of effluent that requires treatment. This environmental efficiency not only reduces disposal costs but also simplifies the regulatory approval process for the manufacturing site. Scalability is further assured by the robustness of the reverse addition hydrolysis, which is less sensitive to minor fluctuations in temperature or mixing rates compared to delicate catalytic couplings, ensuring consistent quality even as batch sizes increase from kilograms to tons.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis of flavonoid intermediates as described in the patent literature. These insights are derived directly from the experimental data and mechanistic explanations provided in CN101570529B, offering clarity on process feasibility and quality control. Understanding these nuances is essential for technical teams evaluating the adoption of this route for their specific pipeline needs. The answers reflect the consensus on best practices for handling sensitive flavonoid structures and optimizing yield.
Q: Why is the reverse addition method critical for this synthesis?
A: Standard hydrolysis fails because the 5-position ester forms a stable intramolecular hydrogen bond with the carbonyl, preventing cleavage. Reverse addition into excess base overcomes this kinetic barrier.
Q: What are the purity advantages of this route over prior art?
A: Unlike previous methods that introduce difficult-to-remove iodine contaminants, this process uses benign acyl protecting groups and standard hydrolysis, ensuring higher clinical safety profiles.
Q: Is this process suitable for large-scale manufacturing?
A: Yes, the route utilizes commercially available starting materials like apigenin and avoids cryogenic conditions or exotic catalysts, making it highly scalable for industrial production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 7-Benzyl-5,4'-dihydroxyapigenin Supplier
The technological advancements detailed in patent CN101570529B underscore the immense potential of flavonoid dimers as next-generation chemosensitizers, yet realizing this potential requires a manufacturing partner with deep technical expertise and scalable capacity. NINGBO INNO PHARMCHEM stands at the forefront of this domain, possessing extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our state-of-the-art facilities are equipped to handle the specific solvent systems and reaction conditions required for this synthesis, ensuring that stringent purity specifications are met consistently. With rigorous QC labs and a commitment to process optimization, we can translate this patented methodology into a robust commercial supply chain that supports your clinical and commercial timelines. We understand that the transition from bench-scale discovery to industrial manufacturing is fraught with challenges, and our team is dedicated to navigating these complexities to deliver high-quality intermediates reliably.
We invite you to engage with our technical procurement team to discuss how this optimized synthetic route can benefit your specific project requirements. By requesting a Customized Cost-Saving Analysis, you can gain a clear understanding of the economic advantages of switching to this high-yield process. We encourage potential partners to contact us for specific COA data and route feasibility assessments tailored to your target molecules. Whether you are in the early stages of drug discovery or preparing for late-stage clinical trials, our expertise in flavonoid chemistry ensures that you have a secure and efficient supply of critical intermediates. Let us collaborate to accelerate the development of life-saving oncology therapies by overcoming the synthetic barriers that have historically hindered progress in this field.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →