Advanced Synthesis of Diazaspiro Intermediates: Technical Upgrade and Commercial Scalability for Global Pharma
Advanced Synthesis of Diazaspiro Intermediates: Technical Upgrade and Commercial Scalability for Global Pharma
The pharmaceutical industry continuously seeks robust synthetic pathways for complex heterocyclic scaffolds that serve as critical building blocks in modern drug discovery, particularly for targets involving metabolic disorders and neurological conditions. Patent CN110922411B discloses a highly efficient synthetic method for 6-(p-toluenesulfonyl)-2,6-diazaspiro[3.3]heptane-6-carboxylic acid tert-butyl ester, a key intermediate with significant potential in the development of active pharmaceutical ingredients. This technical breakthrough addresses long-standing challenges in the synthesis of diazaspiro systems by introducing a concise three-step route that avoids the use of expensive transition metal catalysts and harsh reaction conditions typically associated with prior art methodologies. The innovation lies in the strategic application of nucleophilic substitution and cyclization reactions that proceed under mild thermal conditions, thereby enhancing process safety and operational simplicity for manufacturing teams. By leveraging this patented technology, chemical producers can achieve substantially higher total yields while minimizing the environmental footprint associated with complex multi-step syntheses. This report provides a comprehensive analysis of the technical merits and commercial implications of this novel pathway for stakeholders involved in pharmaceutical intermediate sourcing and process development.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 2,6-diazaspiro[3.3]heptane derivatives has relied on routes that introduce significant operational complexity and cost burdens due to the requirement for specialized reagents and extreme reaction conditions. One conventional pathway involves a sequence of bromination, cyclization, and hydrogenation reduction steps that necessitate the use of palladium on carbon catalysts for the removal of benzyl protecting groups, which not only increases raw material costs but also introduces risks associated with handling pyrophoric catalysts on a large scale. Another existing route utilizes diisobutyl lithium amide (LDA) and requires ultralow temperature conditions around minus 78 degrees Celsius, demanding specialized cryogenic equipment and strictly anhydrous environments that are difficult to maintain consistently in commercial production settings. These traditional methods often suffer from poor operability and low total yields, typically ranging between 15% and 45%, which severely impacts the economic viability of scaling these intermediates for drug development programs. Furthermore, the reliance on hydrogenation steps and harsh reducing agents like lithium aluminum hydride complicates waste treatment and increases the overall safety risk profile of the manufacturing process. The cumulative effect of these limitations is a supply chain that is prone to bottlenecks, higher costs, and longer lead times for critical drug intermediates.
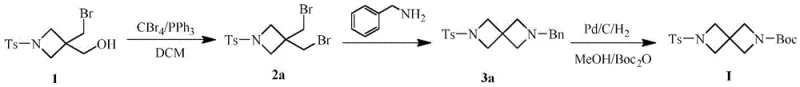
The Novel Approach
In stark contrast to the cumbersome traditional methodologies, the novel approach disclosed in the patent utilizes a streamlined three-step sequence that begins with a mild substitution reaction followed by a halogen exchange and concludes with an efficient cyclization step. This new route strategically avoids the introduction of benzyl protecting groups entirely, thereby eliminating the need for expensive palladium-catalyzed debenzylation and the associated purification challenges that often plague large-scale synthesis. The process operates under significantly milder thermal conditions, with reaction temperatures ranging from 10 to 60 degrees Celsius, which removes the necessity for energy-intensive cryogenic cooling systems and allows for the use of standard glass-lined or stainless steel reactors commonly available in fine chemical facilities. By optimizing the molar ratios of reagents such as triethylamine and metal halides, the method achieves a total yield of approximately 75% to 82%, representing a dramatic improvement over the efficiency of prior art techniques. The simplicity of the workup procedures, which involve standard aqueous washes and extractions, further enhances the practicality of this method for industrial application. This fundamental shift in synthetic strategy offers a compelling value proposition for manufacturers seeking to optimize their production capabilities for high-value pharmaceutical intermediates.
Mechanistic Insights into Substitution and Cyclization
The core of this synthetic innovation lies in the precise control of nucleophilic substitution reactions that transform the starting alcohol into a reactive intermediate capable of undergoing efficient ring closure. In the first step, the hydroxyl group of the starting material is activated through reaction with methanesulfonyl chloride or p-toluenesulfonyl chloride in the presence of an acid-binding agent like triethylamine and a catalyst such as DMAP. This activation converts the poor leaving group hydroxyl into a highly reactive sulfonate ester, facilitating the subsequent displacement by a halide ion in the second step without requiring harsh conditions that might degrade the sensitive spirocyclic core. The use of dimethylaminopyridine as a catalyst accelerates the rate of this substitution reaction while preventing the formation of unwanted chlorinated alkane byproducts, ensuring high selectivity for the desired intermediate. The reaction solvent system, typically dichloromethane or chloroform, is chosen to maintain solubility of the organic substrates while allowing for easy separation of inorganic salts during the workup phase. This careful orchestration of reagents and conditions ensures that the intermediate is generated with high purity, setting the stage for the subsequent cyclization.
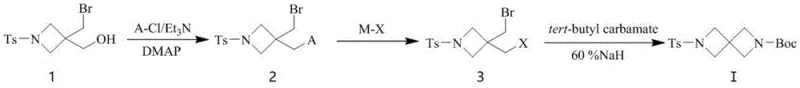
The final cyclization step represents the critical bond-forming event that constructs the diazaspiro[3.3]heptane core, driven by the reaction of the dihalide intermediate with tert-butyl carbamate in the presence of sodium hydride. Sodium hydride acts as a strong base to deprotonate the carbamate, generating a nucleophilic nitrogen species that attacks the electrophilic carbon centers of the dihalide to close the ring system. The reaction is conducted in polar aprotic solvents such as tetrahydrofuran or N,N-dimethylformamide, which stabilize the ionic intermediates and facilitate the displacement of the halide leaving groups. Stirring for not less than half an hour prior to the addition of the dihalide ensures uniform dispersion of the base and complete formation of the nucleophile, which is crucial for maximizing the yield of the cyclization product. The mild heating to 30-60 degrees Celsius provides sufficient activation energy for the ring closure without promoting decomposition or side reactions that could compromise the integrity of the spirocyclic structure. This mechanistic pathway demonstrates a high degree of chemoselectivity, allowing for the formation of the complex spiro framework in a single operational step with minimal impurity generation.
How to Synthesize 6-(p-Toluenesulfonyl)-2,6-Diazaspiro Heptane Ester Efficiently
Implementing this synthesis requires careful attention to reagent quality and reaction monitoring to ensure consistent results across different batch sizes. The process begins with the activation of the starting alcohol, followed by the halogen exchange which converts the sulfonate into a more reactive dihalide species suitable for cyclization. Detailed standard operating procedures for each step, including specific quenching and purification protocols, are essential to maintain the high purity levels required for pharmaceutical applications. The following guide outlines the standardized synthesis steps derived from the patent examples to assist technical teams in replicating this efficient pathway.
- Perform a first substitution reaction on the starting alcohol with methanesulfonyl chloride or p-toluenesulfonyl chloride using triethylamine and DMAP catalyst.
- Conduct a second substitution reaction on the intermediate sulfonate with a metal halide such as lithium bromide or sodium iodide in a polar aprotic solvent.
- Execute a cyclization reaction by adding tert-butyl carbamate and sodium hydride to the dihalide intermediate in tetrahydrofuran or DMF.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain leaders, the adoption of this novel synthetic route translates into tangible strategic advantages that directly impact the bottom line and operational resilience of the organization. By eliminating the need for specialized cryogenic equipment and expensive palladium catalysts, the process significantly reduces the capital expenditure and operational costs associated with manufacturing this critical intermediate. The simplified workflow reduces the number of unit operations required, which in turn minimizes the potential for material loss during transfer and purification stages, leading to better overall mass balance and resource utilization. Furthermore, the use of readily available and stable reagents ensures that the supply chain is less vulnerable to disruptions caused by the scarcity of specialized chemicals often required in complex synthetic routes. This robustness allows for more reliable planning and forecasting, enabling companies to meet tight production schedules without the risk of unexpected delays.
- Cost Reduction in Manufacturing: The elimination of expensive transition metal catalysts and the reduction in energy consumption due to milder reaction temperatures result in substantial cost savings per kilogram of product produced. By avoiding the use of ultralow temperature conditions, facilities can utilize standard reaction vessels without the need for expensive cooling infrastructure, thereby lowering the fixed costs associated with production. The higher total yield achieved by this method means that less raw material is required to produce the same amount of final product, directly improving the cost of goods sold. Additionally, the simplified purification steps reduce the consumption of solvents and chromatography media, further contributing to the overall economic efficiency of the process.
- Enhanced Supply Chain Reliability: The reliance on commodity chemicals such as triethylamine, sodium hydride, and common solvents ensures a stable and diverse supply base that is not dependent on single-source vendors for specialized reagents. This diversification mitigates the risk of supply interruptions and allows for greater flexibility in sourcing strategies, which is critical for maintaining continuous production flows. The robustness of the reaction conditions also means that the process is less sensitive to minor variations in raw material quality, reducing the likelihood of batch failures that can disrupt supply schedules. Consequently, partners can expect more consistent delivery performance and shorter lead times for high-purity pharmaceutical intermediates.
- Scalability and Environmental Compliance: The absence of heavy metal catalysts and harsh reducing agents simplifies the waste treatment process, making it easier to comply with increasingly stringent environmental regulations regarding effluent discharge. The mild reaction conditions facilitate safer scale-up from laboratory to commercial production, as the thermal risks associated with exothermic reactions are easier to manage in large reactors. This scalability ensures that the supply can be rapidly expanded to meet growing market demand without the need for extensive process re-engineering. Moreover, the reduced solvent usage and higher atom economy of the route contribute to a greener manufacturing profile, aligning with corporate sustainability goals.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation and benefits of this synthetic method, based on the detailed data provided in the patent documentation. These answers are designed to clarify the operational advantages and technical feasibility for potential partners evaluating this technology for their supply chains. Understanding these details is crucial for making informed decisions about process adoption and vendor selection.
Q: How does this new synthesis route improve upon conventional methods regarding safety and cost?
A: The novel route eliminates the need for ultralow temperature conditions (-78°C) and expensive palladium-catalyzed hydrogenation steps required in prior art, significantly reducing equipment requirements and operational hazards while improving total yield from approximately 15-45% to over 75%.
Q: What are the key purity and impurity control advantages of this method?
A: By avoiding harsh reducing agents like lithium aluminum hydride and complex protecting group manipulations, the process minimizes side reactions and byproduct formation, facilitating simpler purification workflows and ensuring high-purity specifications suitable for drug substance manufacturing.
Q: Is this synthetic pathway suitable for large-scale commercial production?
A: Yes, the method utilizes mild reaction temperatures (10-60°C) and readily available reagents without requiring specialized high-pressure hydrogenation equipment, making it highly scalable and robust for industrial manufacturing environments.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 6-(p-Toluenesulfonyl)-2,6-Diazaspiro Heptane Ester Supplier
NINGBO INNO PHARMCHEM stands at the forefront of fine chemical manufacturing, leveraging advanced synthetic technologies like the one described in CN110922411B to deliver high-value intermediates to the global pharmaceutical market. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory innovation to industrial reality is seamless and efficient. We maintain stringent purity specifications and operate rigorous QC labs to guarantee that every batch meets the exacting standards required for drug substance manufacturing. Our commitment to technical excellence allows us to optimize these novel routes for maximum yield and cost efficiency, providing our clients with a competitive edge in their own drug development programs.
We invite you to engage with our technical procurement team to discuss how this optimized synthesis can benefit your specific project requirements. By requesting a Customized Cost-Saving Analysis, you can gain detailed insights into the potential economic impact of switching to this more efficient pathway. We encourage you to contact us to obtain specific COA data and route feasibility assessments tailored to your volume needs. Partnering with us ensures access to a reliable supply chain backed by deep technical expertise and a commitment to quality.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →