Advanced Manufacturing of TAF Intermediates: Overcoming Yield and Time Barriers in Antiviral Drug Production
Advanced Manufacturing of TAF Intermediates: Overcoming Yield and Time Barriers in Antiviral Drug Production
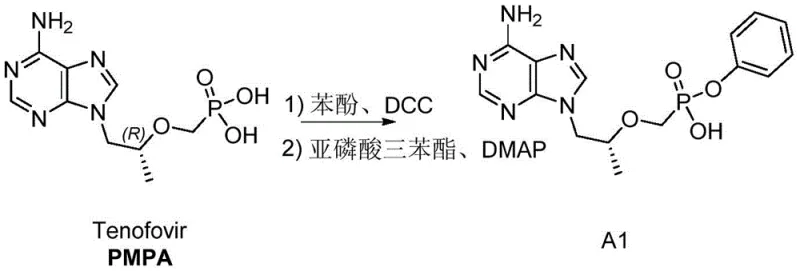
The global demand for potent antiviral therapies, particularly for the treatment of chronic Hepatitis B Virus (HBV) infections, has placed immense pressure on the supply chains of key pharmaceutical intermediates. Tenofovir Alafenamide (TAF), marketed as Vemlidy, represents a significant clinical advancement over its predecessor TDF, offering superior safety profiles regarding renal and skeletal parameters at substantially lower doses. However, the commercial viability of TAF relies heavily on the efficient synthesis of its critical precursors. Patent CN108467410B discloses a groundbreaking preparation method for a specific TAF intermediate, addressing long-standing inefficiencies in yield and reaction time that have plagued previous synthetic routes. This technical insight report analyzes the novel methodology, highlighting its potential to redefine cost structures and supply reliability for reliable TAF intermediate suppliers operating in the competitive antiviral market.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior to the innovations detailed in the referenced patent, the synthesis of TAF intermediates faced significant bottlenecks that hindered efficient cost reduction in pharmaceutical intermediates manufacturing. Traditional routes often relied on phenol-based esterification or the use of triphenyl phosphite in low-boiling solvents like acetonitrile. The phenol route, while chemically feasible, suffered from notoriously low yields, typically hovering around only 60 percent, which is economically unsustainable for large-scale API production. Furthermore, the alternative method utilizing acetonitrile as a solvent presented severe kinetic limitations; the reaction required excessively long durations, often exceeding 72 hours to reach completion, despite some literature suggesting 48 hours. These prolonged reaction times not only tied up reactor capacity but also led to issues with product crystallization and unstable yields, creating unpredictable supply chains for downstream drug manufacturers.
The Novel Approach
The methodology introduced in patent CN108467410B offers a transformative solution by fundamentally altering the reaction environment and workup procedure. By shifting the solvent system to high-boiling aromatic hydrocarbons such as toluene or xylene, the process enables operation at elevated temperatures ranging from 60 to 140 degrees Celsius, with a preferred range of 90 to 110 degrees Celsius. This thermal optimization dramatically accelerates the reaction kinetics, slashing the required reaction time to a mere 10 to 16 hours. Additionally, the introduction of a refined post-treatment protocol involving alkaline washing and specific solvent extractions effectively removes impurities that previously complicated purification. This results in a robust process capable of delivering yields exceeding 90 percent with purity levels greater than 99 percent, establishing a new benchmark for the commercial scale-up of complex pharmaceutical intermediates.
Mechanistic Insights into Triphenyl Phosphite-Mediated Esterification
The core of this synthetic breakthrough lies in the optimized phosphorylation and esterification mechanism mediated by triphenyl phosphite in the presence of catalytic amounts of 4-dimethylaminopyridine (DMAP) and triethylamine. In this system, DMAP acts as a potent nucleophilic catalyst, facilitating the activation of the phosphorus center in triphenyl phosphite, which subsequently attacks the hydroxyl group of the Tenofovir (PMPA) substrate. The use of aromatic solvents is not merely a medium choice but a thermodynamic driver; the higher boiling point of toluene or xylene allows the reaction mixture to maintain a temperature sufficient to overcome the activation energy barrier more rapidly than in acetonitrile. This prevents the accumulation of partially reacted species and drives the equilibrium towards the formation of the desired phenyl phosphate ester bond, ensuring high conversion rates without the need for excessive reagent loading.
Equally critical to the mechanism is the purification strategy which leverages the physicochemical properties of the reaction byproducts. Upon cooling the reaction mixture to between 0 and 50 degrees Celsius, the system undergoes a distinct phase separation where the crude product forms a lower oily layer. This physical separation is followed by an alkaline wash using sodium hydroxide solution, which neutralizes acidic impurities and hydrolyzes residual reactive phosphorus species. The subsequent extraction with ethyl acetate selectively removes organic contaminants while retaining the ionic product in the aqueous phase. Finally, the dropwise addition of concentrated hydrochloric acid lowers the pH, inducing the precipitation of the target intermediate as a crystalline solid. This multi-step purification ensures the removal of trace metals and organic volatiles, crucial for meeting the stringent specifications of high-purity pharmaceutical intermediates.
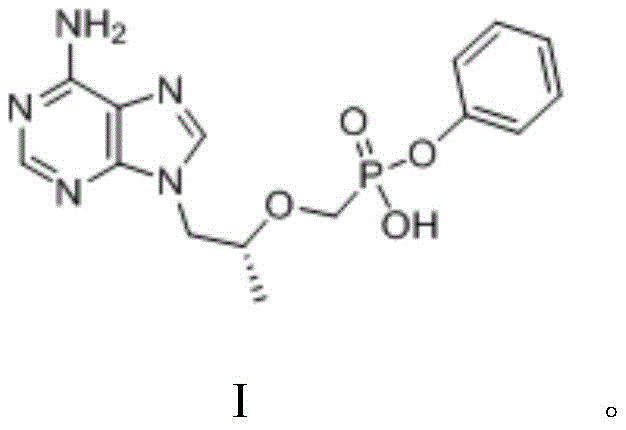
How to Synthesize TAF Intermediate Efficiently
Implementing this novel synthesis route requires precise control over stoichiometry and thermal conditions to maximize the benefits observed in the patent examples. The process begins with the careful charging of PMPA, triphenyl phosphite, DMAP, and triethylamine into a reactor containing the aromatic solvent, followed by a controlled heating ramp to initiate the esterification. The reaction progress is monitored to ensure completion within the 10 to 16-hour window, after which the mixture is cooled to facilitate phase separation. The detailed standardized synthetic steps, including specific mass ratios and crystallization parameters derived from the patent examples, are outlined below to guide process engineers in replicating this high-efficiency protocol.
- Combine PMPA, triphenyl phosphite, DMAP, and triethylamine in toluene or xylene, then heat to 90-140°C for 10-16 hours to complete the esterification reaction.
- Cool the mixture to induce phase separation, collect the lower oily layer, and perform an alkaline wash followed by ethyl acetate extraction to remove impurities.
- Acidify the aqueous phase with concentrated hydrochloric acid to induce crystallization, then filter and dry the solid to obtain the final high-purity intermediate.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented methodology translates directly into enhanced operational resilience and financial efficiency. The primary advantage lies in the drastic reduction of cycle time; by compressing the reaction duration from multiple days to less than a single shift, manufacturers can significantly increase reactor throughput without capital expenditure on new equipment. This acceleration reduces energy consumption per kilogram of product and minimizes the risk of batch failures associated with prolonged heating, thereby stabilizing the supply of this critical antiviral precursor. Furthermore, the simplified workup procedure eliminates the need for complex chromatographic purification, relying instead on scalable liquid-liquid extraction and crystallization techniques that are standard in bulk chemical manufacturing.
- Cost Reduction in Manufacturing: The economic impact of this process is driven by the elimination of inefficient reaction conditions and the optimization of solvent usage. By replacing acetonitrile with recoverable solvents like toluene or xylene, the process reduces raw material costs and simplifies solvent recovery distillation trains. The substantial increase in yield from approximately 60 percent in older phenol routes to over 90 percent in this new method means that less starting material is wasted, directly lowering the cost of goods sold (COGS). Additionally, the reduced reaction time lowers utility costs associated with heating and stirring, contributing to a leaner manufacturing profile that enhances overall profit margins for the final API.
- Enhanced Supply Chain Reliability: Supply continuity is often threatened by processes that are sensitive to minor variations or require hard-to-source reagents. This method utilizes commodity chemicals such as triphenyl phosphite, DMAP, and triethylamine, which are readily available from multiple global suppliers, mitigating the risk of raw material shortages. The robustness of the crystallization step ensures consistent product quality batch-after-batch, reducing the likelihood of out-of-specification results that could delay shipments. This reliability allows supply chain planners to maintain lower safety stock levels while confidently meeting the demanding delivery schedules of major pharmaceutical clients.
- Scalability and Environmental Compliance: From an environmental and scaling perspective, the process is designed to minimize waste generation and simplify effluent treatment. The absence of heavy metal catalysts removes the need for expensive and environmentally taxing metal scavenging steps, aligning with green chemistry principles. The phase separation and extraction workflow is inherently scalable, meaning that the transition from pilot plant kilograms to multi-ton commercial production can be achieved with minimal process re-engineering. This ease of scale-up ensures that manufacturers can rapidly respond to surges in market demand for Tenofovir-based therapies without compromising on environmental regulatory compliance.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this TAF intermediate synthesis route. These answers are derived directly from the experimental data and beneficial effects described in patent CN108467410B, providing clarity on how this technology resolves historical manufacturing pain points. Understanding these details is essential for technical teams evaluating the feasibility of integrating this process into their existing production lines.
Q: How does the new solvent system improve reaction efficiency compared to traditional acetonitrile methods?
A: By utilizing high-boiling aromatic solvents like toluene or xylene instead of acetonitrile, the reaction temperature can be elevated to 90-140°C. This thermal increase drastically accelerates the reaction kinetics, reducing the total process time from over 72 hours in conventional methods to merely 10-16 hours, while simultaneously improving yield stability.
Q: What specific purification steps ensure the high purity (>99%) required for API synthesis?
A: The process incorporates a sophisticated workup sequence involving natural layering to separate the crude oil, followed by a critical alkaline wash to neutralize acidic byproducts. Subsequent extraction with ethyl acetate removes organic impurities, and final crystallization via hydrochloric acid addition ensures the removal of residual salts and unreacted starting materials, achieving purity levels exceeding 99.5%.
Q: Is this synthesis route suitable for large-scale industrial production?
A: Yes, the method is explicitly designed for industrial scalability. It avoids complex chromatographic separations in favor of robust physical separation techniques like phase layering and crystallization. The use of common, recoverable solvents like toluene and the elimination of difficult-to-remove transition metal catalysts make it highly viable for multi-kilogram to ton-scale manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable TAF Intermediate Supplier
As the pharmaceutical industry continues to evolve towards more potent and safer antiviral treatments, the need for high-quality, cost-effective intermediates has never been greater. NINGBO INNO PHARMCHEM stands at the forefront of this evolution, leveraging advanced synthetic methodologies like the one described in CN108467410B to deliver superior value to our global partners. Our facility boasts extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet your volume requirements with consistency. We operate under stringent purity specifications and utilize rigorous QC labs to guarantee that every batch of TAF intermediate meets the highest international standards for safety and efficacy.
We invite you to collaborate with us to optimize your supply chain and reduce your manufacturing costs. Our technical team is prepared to provide a Customized Cost-Saving Analysis tailored to your specific production needs, demonstrating exactly how switching to our optimized route can improve your bottom line. Please contact our technical procurement team today to request specific COA data and route feasibility assessments, and let us help you secure a reliable supply of this critical antiviral building block.