Scalable Manufacturing of CCR-2 Antagonist Intermediates for Global Pharmaceutical Supply Chains
Introduction to Advanced CCR-2 Antagonist Manufacturing
The pharmaceutical industry continuously seeks robust and scalable pathways for complex small molecules, particularly those targeting chemokine receptors like CCR-2. Patent CN101160286B discloses a highly optimized process for the preparation of ((1R,3S)-3-isopropyl-3-{[3-(trifluoromethyl)-7,8-dihydro-1,6-naphthyridin-6(5H)-yl]carbonyl}cyclopentyl)[(3S,4S)-3-methoxytetrahydro-2H-pyran-4-yl]amine and its succinate salt. This technical disclosure represents a significant leap forward from earlier laboratory-scale methods, addressing critical bottlenecks in step count, stereochemical purity, and overall process efficiency. For R&D directors and procurement specialists, understanding this convergent synthesis is vital for securing a reliable supply of high-purity pharmaceutical intermediates. The patent outlines a strategy that not only improves yield but also enhances the physical properties of the final API salt form, ensuring better stability and bioavailability profiles essential for clinical and commercial success.
By leveraging advanced catalytic methodologies and strategic protecting group manipulations, this process transforms what was once a cumbersome multi-step sequence into a streamlined manufacturing protocol. The ability to produce key intermediates such as (3R)-3-methoxytetrahydro-4H-pyran-4-one and 3-(trifluoromethyl)-5,6,7,8-tetrahydro-1,6-naphthyridine with high optical purity is a cornerstone of this technology. As we delve deeper into the technical specifics, it becomes clear that this methodology offers substantial advantages for cost reduction in pharmaceutical manufacturing, making it an attractive candidate for large-scale production by a reliable pharma intermediates supplier.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art techniques for synthesizing this class of CCR-2 antagonists were plagued by inefficiencies that rendered them unsuitable for commercial scale-up. Specifically, the construction of the naphthyridine core traditionally required up to nine separate synthetic steps, involving expensive reagents and generating significant waste. Furthermore, the assembly of the cyclopentene building block often resulted in poor stereocontrol, leading to high proportions of undesired stereoisomers that were difficult and costly to separate. These factors combined to create a process with low overall throughput and high production costs. Additionally, the previously synthesized hydrochloride salt of the target compound exhibited undesirable physicochemical properties, such as high hygroscopicity and poor solubility, which complicated downstream formulation and storage. These limitations underscored the urgent need for a redesigned synthetic route that could deliver both chemical efficiency and superior material properties.
The Novel Approach
The innovative approach detailed in the patent overcomes these historical hurdles through a convergent strategy that couples three distinct building blocks: a functionalized cyclopentene carboxylic acid, a trifluoromethyl-substituted naphthyridine amine, and a chiral methoxy-pyranone. This modular design allows for the independent optimization of each fragment before final assembly. For instance, the naphthyridine fragment is constructed via a concise condensation reaction rather than a lengthy linear sequence. The final assembly involves an amide coupling followed by a highly stereoselective reductive amination. This shift from linear to convergent synthesis drastically reduces the longest linear sequence, thereby improving overall yield and reducing the burden on supply chain logistics. The resulting free base is then converted into a succinate salt, which offers markedly improved stability and processing characteristics compared to the hydrochloride form.
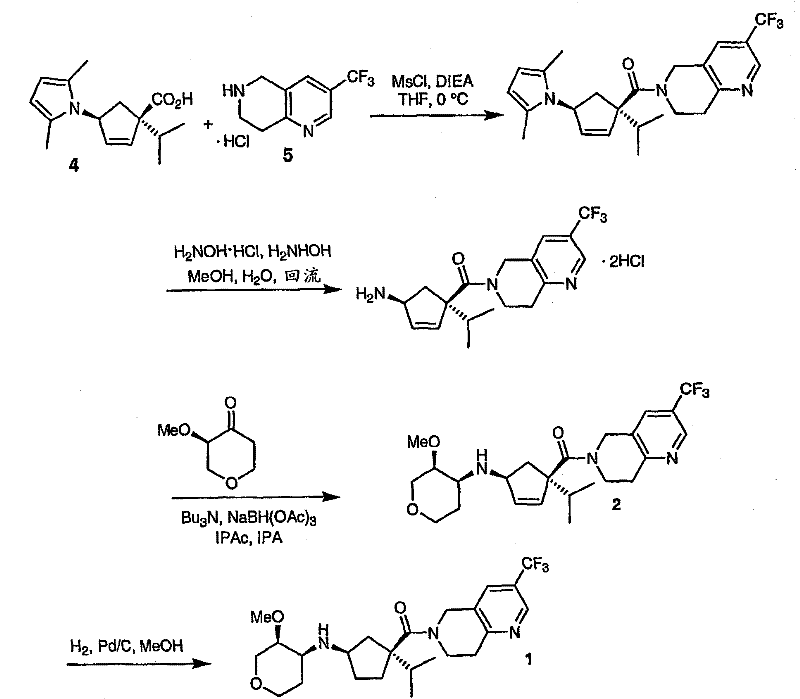
Mechanistic Insights into Asymmetric Dihydroxylation and Reductive Amination
A critical component of this synthesis is the preparation of the chiral pyranone intermediate, (3R)-3-methoxytetrahydro-4H-pyran-4-one. The patent describes a sophisticated application of the Sharpless asymmetric dihydroxylation to install the necessary stereocenters with high fidelity. Starting from a propyl enol ether derived from tetrahydropyran-4-one, the reaction utilizes a chiral ligand system, specifically DHQD2PHAL, in conjunction with osmium tetroxide and NMO as the oxidant. This step typically yields the alpha-hydroxy ketone with an enantiomeric excess (ee) of approximately 80-85%. However, the process does not stop there; it employs a clever purification strategy where the crude product is converted into a bisulfite adduct. By crystallizing this adduct from an acetone-water mixture, the racemic portion is removed, and the mother liquor is enriched to 95-99% ee. Subsequent methylation and deprotection steps preserve this high optical purity, delivering the target pyranone with about 96% ee, which is crucial for the biological activity of the final antagonist.
Another mechanistic highlight is the construction of the cyclopentene fragment, which requires precise control over the quaternary center bearing the isopropyl group. The synthesis begins with a pyrrole-protected aminocyclopentene ester. To introduce the isopropyl group, the process utilizes lithium hexamethyldisilazide (LHMDS) as a strong, non-nucleophilic base to generate the enolate at low temperatures (-20°C). Subsequent alkylation with 2-iodopropane proceeds with high diastereoselectivity, favoring the desired (1S,4S) configuration. The use of the 2,5-dimethylpyrrole protecting group is strategic; it stabilizes the amine during the harsh alkylation conditions and can be subsequently removed under mild acidic or hydroxylamine conditions without affecting the sensitive cyclopentene double bond. This careful orchestration of protecting groups and reaction conditions ensures that the complex stereochemistry of the core ring system is maintained throughout the synthesis, minimizing the formation of impurities that could compromise the quality of the high-purity pharmaceutical intermediates.
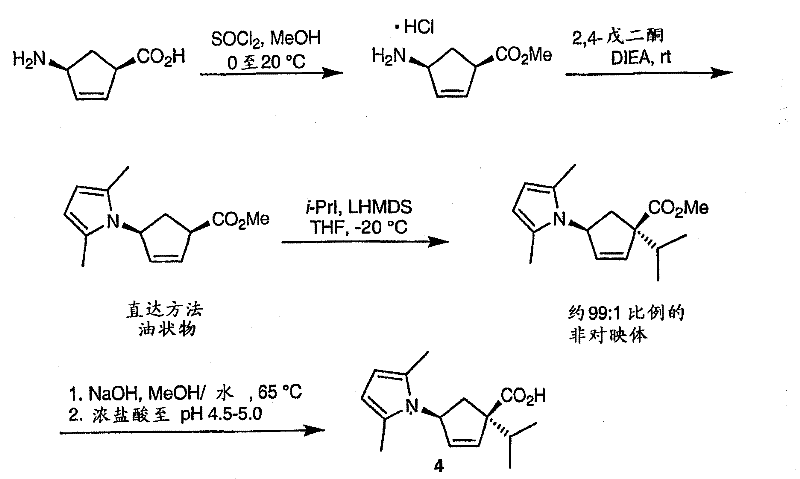
How to Synthesize CCR-2 Antagonist Core Efficiently
The execution of this synthesis requires strict adherence to the specified reaction conditions to ensure reproducibility and safety, particularly when handling reactive intermediates like the vinamidinium salt or strong bases like LHMDS. The process is designed to be telescoped where possible, such as the direct conversion of the amide to the amine salt without isolation, which saves time and solvent. The final hydrogenation step using Pd/C is straightforward but requires careful monitoring to ensure complete reduction of the cyclopentene double bond without over-reduction of other sensitive functionalities. For detailed operational parameters, including specific solvent swaps, temperature ramps, and workup procedures, operators should refer to the standardized protocols derived from the patent examples. These guidelines ensure that the commercial scale-up of complex pharmaceutical intermediates proceeds smoothly from the kilogram to the metric ton scale.
- Prepare the cyclopentene building block via LHMDS-mediated alkylation of a pyrrole-protected aminocyclopentene ester followed by hydrolysis.
- Synthesize the trifluoromethyl-naphthyridine fragment using a vinamidinium salt condensation with protected piperidone.
- Couple the fragments via amide formation, perform reductive amination with the chiral pyranone, and finalize with catalytic hydrogenation.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement perspective, the adoption of this synthetic route offers tangible benefits that extend beyond simple yield improvements. The reduction in step count for the naphthyridine building block directly translates to lower raw material consumption and reduced waste disposal costs. By eliminating the need for nine discrete steps and replacing them with a more direct condensation, the process minimizes the accumulation of impurities and the need for extensive purification between stages. This streamlining effect leads to substantial cost savings in manufacturing, as fewer unit operations mean less energy consumption, lower labor requirements, and reduced equipment occupancy time. Furthermore, the use of readily available starting materials, such as 3,3,3-trifluoropropionic acid and protected piperidones, ensures a stable and resilient supply chain, mitigating the risks associated with sourcing exotic or proprietary reagents.
Supply chain reliability is further enhanced by the robustness of the crystallization processes described for the intermediates. The ability to purify key intermediates like the bisulfite adduct of the pyranone or the succinate salt of the final product through crystallization provides a powerful tool for quality control. Crystallization acts as a purge step, removing trace impurities and ensuring that the final material meets stringent purity specifications without the need for chromatographic separation, which is often difficult to scale. This scalability is a critical factor for supply chain heads who need to guarantee continuous availability of the API for clinical trials and commercial launch. The process is designed to handle large volumes, as evidenced by the kilogram-scale examples provided in the patent, demonstrating that the chemistry is not just theoretically sound but practically viable for industrial production.
Environmental compliance and safety are also significantly improved with this new methodology. The replacement of hazardous reagents and the reduction of solvent usage contribute to a greener manufacturing footprint. The process avoids the use of heavy metal catalysts in the final steps, relying instead on heterogeneous palladium catalysts that can be easily filtered and recycled. This aligns with modern sustainability goals and regulatory expectations for pharmaceutical manufacturing. Additionally, the improved physical properties of the succinate salt form reduce the risks associated with handling hygroscopic materials, enhancing workplace safety and simplifying packaging and storage requirements. These factors collectively make the process not only economically attractive but also environmentally responsible, positioning it as a preferred choice for long-term production strategies.
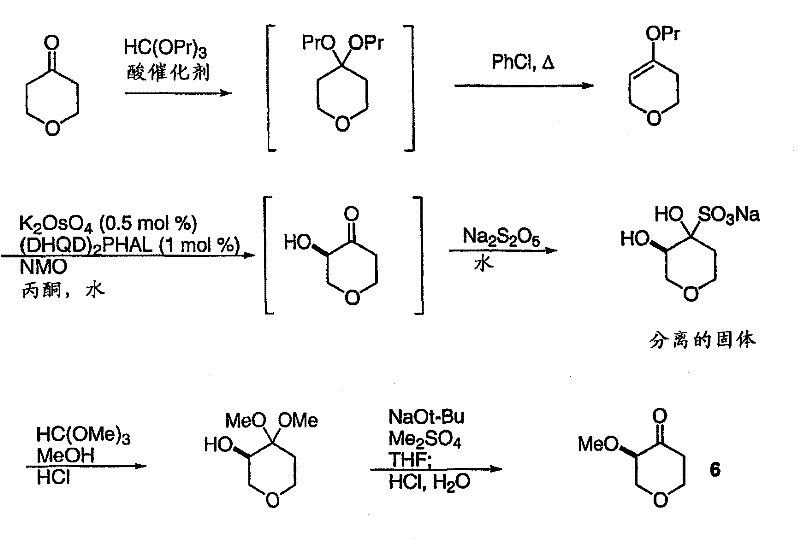
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and optimization of this synthetic route. Understanding these nuances is essential for process chemists and engineers tasked with technology transfer. The answers provided are based on the specific embodiments and data disclosed in the patent literature, offering a realistic view of the process capabilities and limitations. Whether you are evaluating the feasibility of in-house production or assessing the capabilities of a contract manufacturing organization, these insights provide a solid foundation for decision-making regarding the commercialization of this potent CCR-2 antagonist.
Q: What is the key advantage of the new synthesis route for the CCR-2 antagonist?
A: The new route significantly reduces the step count for the naphthyridine building block from nine steps to a more efficient condensation process, while improving stereocontrol in the cyclopentene moiety.
Q: How is high enantiomeric excess achieved in the pyranone intermediate?
A: High ee (up to 99%) is achieved through a modified Sharpless asymmetric dihydroxylation followed by a crystallization-induced diastereomer enrichment of the bisulfite adduct.
Q: Is the succinate salt form more stable than the hydrochloride?
A: Yes, the succinate salt exhibits superior solubility, reduced hygroscopicity, and better processing characteristics compared to the previously synthesized hydrochloride salt.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable CCR-2 Antagonist Supplier
At NINGBO INNO PHARMCHEM, we recognize the complexity and critical nature of synthesizing advanced pharmaceutical intermediates like the CCR-2 antagonist described in CN101160286B. Our team of expert process chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project transitions seamlessly from the lab to the plant. We are committed to delivering materials that meet stringent purity specifications, utilizing our rigorous QC labs to verify every batch against the highest industry standards. Our facility is equipped to handle the specific challenges of this synthesis, including the safe management of reactive intermediates and the precise control required for asymmetric transformations.
We invite you to collaborate with us to optimize your supply chain for this high-value therapeutic candidate. By partnering with NINGBO INNO PHARMCHEM, you gain access to a Customized Cost-Saving Analysis tailored to your specific volume requirements and timeline. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments. Let us demonstrate how our technical expertise and manufacturing capacity can support your goal of bringing this innovative medicine to patients faster and more efficiently.