Advanced Synthesis of Alkannin Acetyl Glucose Derivatives for Commercial Anti-Tumor Applications
Introduction to Novel Alkannin Glycoside Technology
The pharmaceutical landscape is constantly evolving towards more potent and selective anti-tumor agents, and Patent CN101671376B represents a significant breakthrough in the modification of natural naphthoquinones. This intellectual property discloses a robust and efficient preparation method for alkannin acetyl glucose derivatives, a class of compounds demonstrating superior biological activity compared to the parent molecule, Shikonin. The core innovation lies in a streamlined four-step synthetic route that transforms readily available natural sugars into high-value glycosyl donors, which are then coupled with the Shikonin aglycone under strictly controlled cryogenic conditions. This approach not only enhances the water solubility and bioavailability of the therapeutic agent but also significantly improves its efficacy against drug-resistant tumor cell lines. By leveraging specific Lewis acid catalysis and protecting group strategies, the technology ensures the precise formation of the beta-glycosidic bond, a critical structural feature for maintaining biological potency.
For procurement specialists and supply chain managers, understanding the underlying chemistry is vital for assessing long-term viability. The described methodology moves away from erratic extraction-dependent supplies of rare natural derivatives and establishes a reproducible semi-synthetic pathway. This shift is paramount for securing a reliable pharmaceutical intermediates supplier relationship, as it decouples production from agricultural variables. The patent highlights the versatility of the method, accommodating a wide range of monosaccharides and disaccharides, including glucose, mannose, galactose, and even complex disaccharides like maltose and lactose. This modularity suggests a platform technology capable of generating diverse libraries of analogues for structure-activity relationship (SAR) studies, thereby de-risking the R&D pipeline for potential licensees and partners seeking next-generation oncology therapeutics.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the chemical modification of Shikonin and its analogues has been plagued by significant challenges related to regioselectivity and stability. Traditional esterification or etherification methods often require harsh acidic or basic conditions that can degrade the sensitive 5,8-dihydroxy-1,4-naphthoquinone chromophore, leading to complex impurity profiles and reduced yields. Furthermore, direct glycosylation attempts frequently result in poor stereocontrol, producing mixtures of alpha and beta anomers that are difficult and costly to separate on a large scale. The presence of multiple hydroxyl groups on both the sugar and the aglycone necessitates extensive protecting group manipulation, which traditionally involves toxic heavy metal catalysts or expensive reagents that complicate waste disposal and increase the overall cost of goods sold (COGS). These inefficiencies create bottlenecks in cost reduction in API manufacturing, making many promising Shikonin derivatives commercially unviable despite their potent in vitro activity.
The Novel Approach
The methodology outlined in the patent data introduces a sophisticated solution through the use of trichloroacetimidate glycosyl donors, a strategy renowned for its efficiency and mildness in carbohydrate chemistry. As illustrated in the reaction scheme below, the process begins with the peracetylation of natural sugars, followed by a highly selective deprotection at the anomeric position to generate a hemiacetal. This intermediate is then converted into a trichloroacetimidate donor, which serves as a highly reactive yet stable precursor for glycosylation. The coupling reaction with Shikonin is catalyzed by boron trifluoride diethyl etherate (BF3·Et2O) at low temperatures ranging from -20°C to -40°C. This cryogenic environment is crucial as it kinetically favors the formation of the beta-anomer while suppressing side reactions. The inclusion of 4 Angstrom molecular sieves further drives the reaction to completion by scavenging trace moisture, ensuring high conversion rates and simplifying downstream purification.
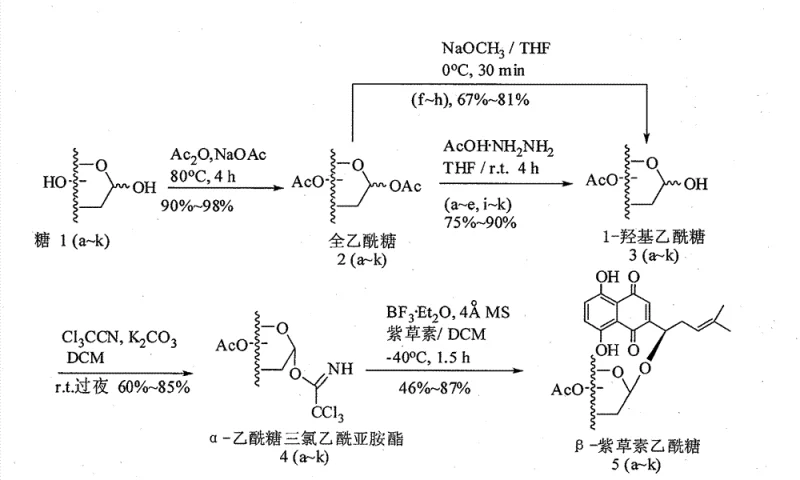
This novel approach offers distinct advantages for commercial scale-up of complex pharmaceutical intermediates. By avoiding harsh conditions, the integrity of the naphthoquinone core is preserved, leading to cleaner reaction profiles and higher purity final products. The use of acetic anhydride and common organic solvents like dichloromethane and tetrahydrofuran ensures that raw material costs remain low and supply chains are robust. Moreover, the stereoselectivity achieved eliminates the need for costly chiral separations, directly impacting the bottom line. For a reliable pharmaceutical intermediates supplier, this translates to a process that is not only chemically elegant but also economically sound, capable of delivering high-purity alkannin derivatives consistently across multiple batches.
Mechanistic Insights into BF3·Et2O Catalyzed Glycosylation
The success of this synthesis hinges on the precise mechanistic interplay between the trichloroacetimidate donor and the Lewis acid catalyst. When BF3·Et2O is introduced to the reaction mixture at -40°C, it coordinates with the nitrogen atom of the trichloroacetimidate group, increasing the electrophilicity of the anomeric carbon. This activation facilitates the departure of the trichloroacetamide leaving group, generating a transient oxocarbenium ion intermediate. The Shikonin molecule, acting as the glycosyl acceptor, attacks this electrophilic center. The low temperature is critical here; it slows down the reaction kinetics enough to allow the neighboring group participation of the C-2 acetyl group (in the case of glucose, mannose, and galactose derivatives) to direct the incoming nucleophile from the opposite face, thereby ensuring the exclusive formation of the 1,2-trans (beta) glycosidic linkage. This level of control is essential for maintaining the biological activity of the final drug candidate.
Furthermore, the reaction environment is meticulously managed to prevent hydrolysis of the sensitive imidate donor. The patent specifies the use of activated 4 Angstrom molecular sieves, which act as a desiccant to remove any trace water that might otherwise quench the oxocarbenium ion or hydrolyze the donor back to the hemiacetal. This attention to detail in moisture control is a hallmark of robust process chemistry. Following the coupling, the reaction is quenched with triethylamine to neutralize the Lewis acid, followed by acidification with acetic acid to induce a color change indicative of the product's stability. This workup procedure is designed to be operationally simple, allowing for filtration and solvent removal without requiring exotic extraction techniques. Such mechanistic clarity provides R&D directors with confidence in the reproducibility of the process, ensuring that the high-purity alkannin derivatives produced meet stringent regulatory specifications for clinical evaluation.
How to Synthesize Alkannin Acetyl Glucose Efficiently
The synthesis of these valuable anti-tumor intermediates follows a logical progression designed to maximize yield and stereochemical purity. The process initiates with the protection of the sugar hydroxyl groups, rendering them inert to subsequent reaction conditions while activating the anomeric center for coupling. The transformation of the protected sugar into a trichloroacetimidate donor is a pivotal step that converts a stable carbohydrate into a potent glycosylating agent. Finally, the coupling with Shikonin under Lewis acid catalysis stitches the two pharmacophores together. The detailed standardized synthesis steps for implementing this technology in a GMP environment are provided in the guide below.
- Peracetylation of natural sugars using acetic anhydride and sodium acetate at 80°C to form fully protected sugar donors.
- Selective removal of the anomeric acetyl group using hydrazine acetate or sodium methoxide to generate 1-hydroxy sugars.
- Conversion to trichloroacetimidate donors using trichloroacetonitrile and potassium carbonate, followed by coupling with Shikonin using BF3·Et2O at -40°C.
Commercial Advantages for Procurement and Supply Chain Teams
From a strategic sourcing perspective, the adoption of this synthetic route offers substantial benefits for reducing supply chain vulnerability and optimizing manufacturing costs. Traditional methods of obtaining Shikonin derivatives often rely on plant extraction, which is subject to seasonal variations, geographical constraints, and fluctuating potency. By shifting to a semi-synthetic approach using commodity sugars, manufacturers can secure a consistent and scalable supply of raw materials. This transition effectively mitigates the risk of raw material shortages and price volatility, ensuring enhanced supply chain reliability for downstream drug development projects. The ability to synthesize a library of analogues (from 5a to 5k) from a common set of precursors further allows for inventory optimization, where a single facility can produce multiple SKUs without significant retooling.
- Cost Reduction in Manufacturing: The elimination of expensive transition metal catalysts and the use of inexpensive, bulk-available reagents like acetic anhydride and sodium acetate significantly lowers the direct material costs. Furthermore, the high stereoselectivity of the glycosylation step reduces the burden on purification processes, minimizing solvent consumption and waste generation. This streamlined workflow translates to a lower cost per kilogram of the active intermediate, providing a competitive edge in cost reduction in API manufacturing.
- Enhanced Supply Chain Reliability: The reliance on naturally occurring sugars such as glucose, galactose, and ribose ensures that the starting materials are sourced from stable, global supply chains. Unlike rare natural extracts, these commodities are produced in massive quantities for the food and fermentation industries, guaranteeing availability. This stability allows for long-term supply agreements and reduces the lead time associated with sourcing specialized botanical extracts, thereby reducing lead time for high-purity pharmaceutical intermediates.
- Scalability and Environmental Compliance: The reaction conditions, while requiring low temperatures, are fully compatible with standard stainless steel jacketed reactors used in fine chemical production. The process avoids the generation of heavy metal waste, simplifying environmental compliance and wastewater treatment. The scalability of the chromatography purification steps, combined with the high yields reported in the patent embodiments, supports the commercial scale-up of complex pharmaceutical intermediates from pilot plant to multi-ton annual production without compromising quality.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of alkannin acetyl glucose derivatives. These insights are derived directly from the experimental data and claims within the patent documentation, providing a transparent view of the technology's capabilities. Understanding these details is crucial for stakeholders evaluating the feasibility of integrating these intermediates into their oncology drug pipelines.
Q: What is the primary advantage of the trichloroacetimidate method for Shikonin glycosylation?
A: The trichloroacetimidate method allows for mild, Lewis acid-catalyzed coupling at low temperatures (-20°C to -40°C), which ensures high beta-stereoselectivity and prevents degradation of the sensitive naphthoquinone core of Shikonin.
Q: How does this synthesis address drug resistance in tumor cells?
A: The resulting alkannin acetyl glucose derivatives exhibit strong cytotoxic activity against multi-drug resistant (MDR) cell lines such as K562/Adr and MCF-7/Adr, effectively reversing resistance mechanisms compared to native Shikonin.
Q: Is this process scalable for industrial production?
A: Yes, the process utilizes commodity starting materials like natural sugars and standard reagents (acetic anhydride, BF3·Et2O). The reaction conditions are compatible with standard jacketed reactors, facilitating scale-up from laboratory to commercial tonnage.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Alkannin Acetyl Glucose Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of the synthetic route described in Patent CN101671376B for developing next-generation anti-cancer therapeutics. Our team of process chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to industrial reactor is seamless and efficient. We are committed to delivering high-purity alkannin derivatives that meet the rigorous demands of global pharmaceutical clients, supported by our state-of-the-art analytical capabilities and stringent purity specifications. Our rigorous QC labs utilize advanced chromatography and spectroscopy to verify the stereochemical integrity and chemical purity of every batch, guaranteeing that the material you receive is ready for formulation or further synthesis.
We invite you to collaborate with us to optimize your supply chain for these critical oncology intermediates. Our technical procurement team is prepared to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating how our manufacturing efficiencies can lower your overall project costs. We encourage you to contact us to request specific COA data and route feasibility assessments for any of the alkannin acetyl glucose analogues (5a-5k). Let us be your partner in bringing these promising anti-tumor agents from concept to clinic.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →