Advanced Synthesis of TGF-β1 Targeted Degradation Compounds for Commercial Pharma Applications
The pharmaceutical industry is currently witnessing a paradigm shift from traditional inhibition strategies to targeted protein degradation, a field revolutionized by Proteolysis Targeting Chimeras (PROTACs). Patent CN110862434A discloses a groundbreaking class of compounds capable of promoting the targeted degradation of Transforming Growth Factor-beta 1 (TGF-β1), a cytokine critically involved in fibrosis and tumorigenesis. Unlike conventional small molecule inhibitors that merely block active sites, these bifunctional molecules recruit the E3 ubiquitin ligase Cereblon (CRBN) to the TGF-β1 protein, forcing its destruction via the ubiquitin-proteasome pathway. This technological leap offers a potent solution for treating cancers, tissue fibrosis, and skin scarring where TGF-β1 overexpression is a pathological hallmark. For R&D directors and procurement specialists, understanding the synthetic accessibility and structural modularity of these agents is paramount for securing a reliable supply chain for next-generation therapeutics.
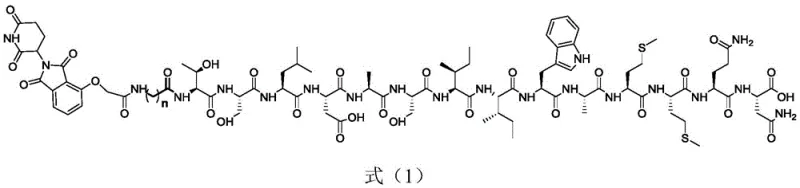
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, therapeutic strategies targeting TGF-β signaling have relied heavily on antisense oligonucleotides, monoclonal antibodies, or small molecule kinase inhibitors. While these approaches have shown promise in preclinical models, they face significant clinical hurdles that limit their widespread adoption and commercial viability. Antibodies often suffer from poor tissue penetration and high manufacturing costs, while small molecule inhibitors frequently encounter issues with drug resistance due to mutation of the kinase domain or compensatory pathway activation. Furthermore, simply inhibiting the signaling pathway does not reduce the overall burden of the TGF-β1 protein itself, which may continue to exert non-canonical effects or accumulate in the extracellular matrix. These limitations create an urgent demand for novel mechanisms that can permanently remove the pathogenic protein rather than just transiently blocking its function, driving the industry towards degradation-based modalities.
The Novel Approach
The technology described in the patent introduces a heterobifunctional molecule design that elegantly overcomes the shortcomings of prior art by utilizing the cell's own waste disposal machinery. By conjugating a TGF-β1 binding peptide to a thalidomide-derived CRBN ligand via a tunable alkyl linker, the compound acts as a molecular bridge. This proximity-induced degradation ensures high specificity and catalytic turnover, meaning a single molecule can degrade multiple targets over time. From a manufacturing perspective, this approach leverages well-established solid-phase peptide synthesis (SPPS) techniques combined with small molecule organic synthesis. This hybrid strategy allows for rapid optimization of the linker length and peptide sequence without requiring entirely new synthetic routes, thereby accelerating the lead optimization phase and reducing the time-to-market for potential drug candidates targeting fibrotic diseases and oncology indications.
Mechanistic Insights into Thalidomide-Based PROTAC Assembly
The core mechanism relies on the precise spatial arrangement of three distinct functional domains within a single molecular entity. The first domain is a 14-amino acid peptide sequence specifically engineered to bind with high affinity to the TGF-β1 protein. The second domain is a thalidomide derivative, which serves as the ligand for the E3 ubiquitin ligase Cereblon (CRBN). The third component is an alkyl linker that connects these two warheads with the optimal length and flexibility to allow the formation of a stable ternary complex. Once inside the cell, the compound binds TGF-β1 and recruits CRBN simultaneously. This event triggers the transfer of ubiquitin molecules onto lysine residues of the TGF-β1 protein. Polyubiquitinated TGF-β1 is then recognized by the 26S proteasome and degraded into small peptides, effectively lowering the intracellular and secreted levels of the cytokine. This catalytic mode of action distinguishes it from stoichiometric inhibitors and offers a profound advantage in potency and durability of response.
Controlling impurities in such complex bifunctional molecules is critical for ensuring safety and efficacy. The synthesis involves coupling sensitive peptide sequences with small molecule warheads, which can lead to deletion sequences, racemization, or incomplete couplings. The patent specifies the use of Fmoc/tBu protection strategies, which are orthogonal and mild, minimizing side reactions during chain elongation. Furthermore, the final cleavage step utilizes a specific cocktail of trifluoroacetic acid (TFA) with scavengers like phenol and triisopropylsilane to prevent alkylation of sensitive residues like tryptophan or methionine. Rigorous purification via reversed-phase high-performance liquid chromatography (RP-HPLC) is employed to isolate the target compound with high purity, ensuring that the final product meets the stringent specifications required for clinical-grade pharmaceutical intermediates and reducing the risk of immunogenic responses caused by peptide impurities.
How to Synthesize TGF-β1 Degrader Intermediates Efficiently
The preparation of these high-value compounds follows a convergent synthesis strategy that maximizes yield and purity while minimizing operational complexity. The process begins with the solution-phase synthesis of the thalidomide derivative warhead, followed by its conjugation to a growing peptide chain on a solid support. This hybrid approach combines the robustness of small molecule chemistry for the warhead with the automation potential of SPPS for the peptide domain. The versatility of the linker region allows for the generation of a homologous series of compounds (e.g., DT-6, DT-7, DT-8) by simply varying the amino acid or alkyl chain length during the synthesis. Detailed standardized synthetic protocols are essential for reproducibility, particularly when scaling up from milligram research quantities to kilogram commercial production. The following guide outlines the critical stages of this sophisticated manufacturing process.
- Synthesize the thalidomide derivative warhead by reacting 3-hydroxyphthalic anhydride with 3-aminopyridine-2,6-dione hydrochloride, followed by alkylation and deprotection to obtain the free acid linker.
- Perform standard Fmoc/tBu solid-phase peptide synthesis on o-chlorotrityl chloride resin, sequentially coupling Fmoc-protected amino acids to build the 14-peptide TGF-β1 binding domain.
- Conjugate the alkyl linker and the pre-synthesized thalidomide derivative to the resin-bound peptide, followed by global cleavage using TFA/scavengers and purification via RP-HPLC.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this synthetic route offers substantial strategic benefits over traditional biologic or complex small molecule manufacturing. The reliance on standard Fmoc-protected amino acids and commercially available thalidomide precursors ensures a robust and diversified supply base, mitigating the risk of raw material shortages. Unlike biologics which require cold chain logistics and fermentation facilities, these peptide-small molecule hybrids can be produced using standard chemical manufacturing infrastructure. This compatibility with existing fine chemical plants significantly lowers the barrier to entry for contract development and manufacturing organizations (CDMOs), fostering a competitive supplier landscape that drives down costs. Additionally, the modular nature of the synthesis allows for rapid adjustment of production volumes based on demand forecasts without extensive retooling.
- Cost Reduction in Manufacturing: The elimination of transition metal catalysts and the use of straightforward coupling reagents like HBTU and HOBt streamline the production process, removing the need for expensive and technically demanding metal scavenging steps. This simplification directly translates to lower operational expenditures and reduced waste treatment costs associated with heavy metal disposal. Furthermore, the high efficiency of solid-phase synthesis minimizes solvent consumption per unit of product compared to multi-step solution phase peptide synthesis, contributing to a more sustainable and cost-effective manufacturing profile that aligns with green chemistry initiatives.
- Enhanced Supply Chain Reliability: The starting materials, including 3-hydroxyphthalic anhydride and standard Fmoc-amino acids, are commodity chemicals available from multiple global suppliers. This diversity in sourcing prevents single-point failures in the supply chain and ensures business continuity even during market fluctuations. The synthetic route avoids the use of exotic or highly regulated precursors that often face export controls or long lead times, thereby reducing the overall lead time for high-purity pharmaceutical intermediates and enabling faster response to clinical trial material requests.
- Scalability and Environmental Compliance: The process is inherently scalable due to the linear nature of solid-phase synthesis, which can be easily adapted from manual reactors to automated synthesizers and large-scale columns. The waste streams are primarily organic solvents and salts, which are well-understood and manageable within standard environmental compliance frameworks. The absence of biological fermentation byproducts simplifies the purification train and reduces the environmental footprint, making it easier for manufacturers to meet increasingly strict regulatory standards for pharmaceutical production while maintaining high throughput capabilities.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these TGF-β1 degraders. These insights are derived directly from the patent specifications and are intended to clarify the feasibility of integrating these intermediates into your drug discovery pipeline. Understanding the nuances of the synthesis and mechanism is crucial for making informed decisions about partnering with suppliers and planning development timelines.
Q: What is the primary mechanism of action for these TGF-β1 degraders?
A: These compounds function as PROTACs (Proteolysis Targeting Chimeras) that simultaneously bind to the TGF-β1 protein and the E3 ubiquitin ligase Cereblon (CRBN). This ternary complex formation induces the ubiquitination of TGF-β1, marking it for degradation by the intracellular ubiquitin-proteasome system, thereby reducing pathological levels of the cytokine.
Q: How does the solid-phase synthesis approach impact production scalability?
A: The use of standard Fmoc/tBu solid-phase peptide synthesis (SPPS) allows for highly automated and reproducible manufacturing. This methodology eliminates the need for complex intermediate isolations during chain elongation, significantly streamlining the process and facilitating easier scale-up from laboratory to commercial production volumes compared to traditional solution-phase peptide synthesis.
Q: Are these intermediates suitable for developing anti-fibrotic therapies?
A: Yes, the patent data explicitly demonstrates that these compounds effectively degrade TGF-β1 in HepG2 cells and inhibit its secretion in M2 macrophages. Since TGF-β1 is a key driver of tissue fibrosis and tumor progression, these intermediates are highly relevant for developing novel therapeutics targeting fibrosis, cancer, and skin scarring.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable TGF-β1 Degrader Supplier
The development of targeted protein degraders represents one of the most exciting frontiers in modern medicine, yet translating these complex molecules from bench to bedside requires a partner with deep technical expertise and proven scale-up capabilities. NINGBO INNO PHARMCHEM stands at the forefront of this innovation, offering comprehensive CDMO services tailored to the unique challenges of PROTAC and peptide-drug conjugate manufacturing. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can seamlessly transition from preclinical research to late-stage clinical trials and beyond. We maintain stringent purity specifications and operate rigorous QC labs equipped with state-of-the-art analytical instrumentation to guarantee the quality and consistency of every batch.
We invite you to collaborate with us to accelerate your fibrosis and oncology programs. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis that evaluates your specific route requirements and identifies opportunities for process optimization. Contact us today to request specific COA data for our reference standards and discuss detailed route feasibility assessments. Let us be your strategic partner in bringing these transformative TGF-β1 degradation therapies to patients worldwide.