Advanced Synthetic Route for 6 Alpha 9 Alpha Diflupredone: Commercial Scalability and Purity
The pharmaceutical industry continuously seeks robust synthetic pathways for potent corticosteroids, and the preparation method detailed in patent CN110964076B represents a significant leap forward in the manufacturing of 6 alpha, 9 alpha-diflupredone. This specific compound serves as a critical active pharmaceutical ingredient (API) or advanced intermediate for anti-inflammatory medications, notably ophthalmic emulsions approved by the FDA for treating post-operative inflammation. The disclosed technology utilizes anecortave dehydroacetate as a strategic starting material, diverging from traditional prednisolone-based routes to offer a streamlined process characterized by bromine hydroxyl epoxy formation, site-specific fluorination at the 6 and 9 positions, and final hydrolysis. For R&D directors and procurement specialists evaluating reliable hormone suppliers, this patent offers a compelling value proposition through its ability to minimize isomeric impurities and shorten the overall reaction sequence. By shifting the synthetic entry point to anecortave derivatives, the process inherently reduces the complexity of protecting group manipulations that have historically plagued steroid synthesis, thereby establishing a new benchmark for efficiency in steroid hormone preparation.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of difluorinated corticosteroids like difluprednate has relied on complex multi-step sequences that introduce significant operational risks and cost inefficiencies. As illustrated in prior art such as route one from patent ZA6803686, conventional methodologies typically involve early-stage esterification of hydroxyl groups at positions 17 and 21, followed by the formation of epoxy groups at positions 9 and 11 under alkaline conditions. This sequence is fundamentally flawed because the alkaline environment required for epoxidation inevitably triggers the hydrolysis of the previously installed ester groups, leading to a cascade of side reactions and the generation of difficult-to-remove impurities. Furthermore, these traditional routes often necessitate the separation of intermediate states, which drastically increases solvent consumption, processing time, and material loss. The reliance on harsh oxidants like perchloric acid in some legacy methods also introduces severe safety hazards, including the risk of combustion when contacting organic substances, which complicates regulatory compliance and insurance for manufacturing facilities. Consequently, the cumulative effect of these inefficiencies results in lower overall yields and a higher cost of goods sold (COGS), making the final API less competitive in the global market.
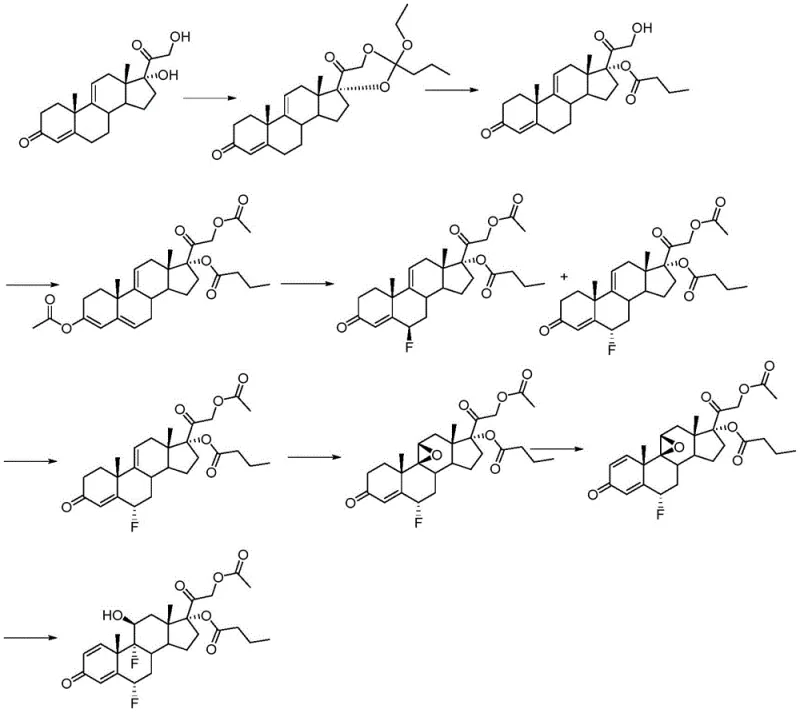
The Novel Approach
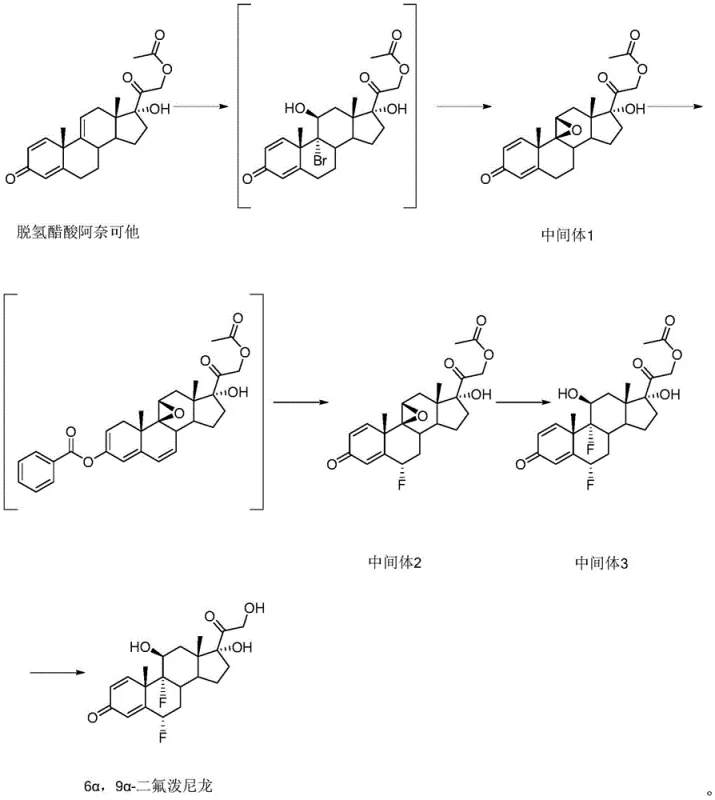
In stark contrast to the convoluted legacy pathways, the novel approach described in CN110964076B leverages anecortave dehydroacetate to bypass many of these thermodynamic and kinetic bottlenecks. This innovative route strategically delays certain functional group manipulations, allowing for a more linear and controlled progression towards the target molecule. By initiating the synthesis with a substrate that already possesses a favorable oxidation state and structural conformation, the process eliminates the need for redundant protection and deprotection cycles that characterize older methods. The implementation of a one-pot method for the fluorination steps further exemplifies this efficiency, where intermediate compounds are not isolated but directly subjected to selective fluorine reagents, thereby minimizing material handling and exposure to environmental contaminants. This streamlined architecture not only enhances the chemical purity of the crude product but also significantly simplifies the downstream purification processes, such as crystallization and filtration. For supply chain heads, this translates to a more predictable manufacturing timeline and reduced dependency on scarce reagents, ensuring a stable supply of high-purity pharmaceutical intermediates.
Mechanistic Insights into Fluoroboric Acid Catalyzed Epoxidation
A critical differentiator in this patented technology is the substitution of perchloric acid with fluoroboric acid during the formation of the bromohydrin intermediate, a change that profoundly impacts both safety and product quality. Mechanistically, fluoroboric acid acts as a milder yet effective catalyst that facilitates the electrophilic addition of bromine across the double bond without inducing the aggressive oxidative degradation associated with perchloric acid. This subtle shift in reagent choice prevents the formation of oxidation by-products that are notoriously difficult to separate from the steroid backbone, thereby preserving the integrity of the molecular structure throughout the synthesis. The reaction is conducted at a controlled temperature range of 0-5 ℃, which is essential for maintaining the stereochemical fidelity of the epoxide ring formation. Following the bromination, the intermediate is treated with an alkaline solution, preferably potassium carbonate, at 35-45 ℃ to induce cyclization. This specific thermal window ensures complete conversion to the epoxy intermediate while preventing the thermal decomposition of the sensitive steroid nucleus. The result is a robust intermediate with a purity exceeding 97%, providing a solid foundation for subsequent fluorination steps.
Furthermore, the control of impurity profiles is meticulously managed through the use of benzoyl chloride for the esterification of the 3-carbonyl group prior to fluorination. This protective step is crucial because it inhibits the generation of isomers during the subsequent reaction with selective fluorine reagents, a common pitfall in non-protected syntheses. The process employs a selective fluorine reagent (CAS 140681-55-6) in an acetonitrile and pyridine solvent system under a nitrogen atmosphere, ensuring that fluorination occurs exclusively at the desired 6-position without affecting other sensitive sites. The subsequent 9-position fluorination utilizes hydrofluoric acid at cryogenic temperatures (-10 to -5 ℃), a condition that demands precise engineering controls but yields a product with minimal side reactions. The inclusion of antioxidants such as tert-butyl hydroquinone during the workup phase further safeguards the product against oxidative degradation, ensuring that the final 6 alpha, 9 alpha-diflupredone meets stringent purity specifications of greater than 98%. This rigorous attention to mechanistic detail underscores the feasibility of the process for large-scale commercial production.
How to Synthesize 6 Alpha 9 Alpha Diflupredone Efficiently
The synthesis of 6 alpha, 9 alpha-diflupredone via this patented route involves a sequence of highly controlled chemical transformations designed to maximize yield and safety. The process begins with the dissolution of anecortave dehydroacetate in a solvent mixture, followed by the dropwise addition of fluoroboric acid and dibromohydantoin at low temperatures to form the bromohydrin precursor. Subsequent steps involve careful pH adjustment and temperature modulation to achieve cyclization, followed by the critical fluorination stages which require inert atmospheres and specific reagent stoichiometry. The final hydrolysis step releases the active hydroxyl groups, yielding the target molecule after purification. While the general chemical logic is straightforward, the precise execution requires adherence to strict operational parameters regarding temperature, mixing rates, and quenching protocols to ensure reproducibility. The detailed standardized synthesis steps, including specific reagent quantities and processing times, are outlined in the guide below for technical reference.
- React anecortave dehydroacetate with dibromohydantoin and fluoroboric acid to form the bromohydrin intermediate, followed by cyclization to the epoxy compound.
- Perform selective 6-position fluorination using a specialized fluorine reagent in acetonitrile and pyridine under nitrogen protection.
- Execute 9-position fluorination using hydrofluoric acid at low temperatures, followed by alkaline hydrolysis to yield the final product.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this synthesis route offers substantial strategic advantages for procurement managers and supply chain leaders focused on cost reduction in pharmaceutical manufacturing. The primary driver of value is the significant simplification of the process flow, which directly correlates to reduced operational expenditures (OPEX) and capital expenditures (CAPEX). By eliminating the need for intermediate isolation and reducing the total number of unit operations, manufacturers can achieve faster batch cycle times and lower utility consumption. The switch to safer reagents like fluoroboric acid also mitigates the need for specialized explosion-proof infrastructure, further lowering the barrier to entry for production and reducing insurance premiums. Additionally, the high purity of the crude product minimizes the load on purification systems, extending the lifespan of chromatography columns and reducing solvent waste disposal costs. These factors collectively contribute to a more resilient and cost-effective supply chain capable of meeting the demanding volume requirements of the global ophthalmic drug market.
- Cost Reduction in Manufacturing: The elimination of expensive transition metal catalysts and the reduction in solvent usage through one-pot methodologies lead to substantial cost savings. By avoiding the hydrolysis of ester groups seen in conventional routes, the process prevents the loss of valuable starting materials, thereby improving the overall mass balance and reducing the cost per kilogram of the final API. The use of readily available raw materials like anecortave dehydroacetate ensures stable pricing and reduces exposure to volatile commodity markets.
- Enhanced Supply Chain Reliability: The robustness of this synthetic route enhances supply chain continuity by reducing the risk of batch failures due to impurity buildup. The shorter reaction sequence means fewer potential points of failure, allowing for more consistent delivery schedules and reduced lead times for high-purity hormone intermediates. Furthermore, the safety improvements associated with replacing perchloric acid reduce the likelihood of regulatory shutdowns or safety incidents that could disrupt production.
- Scalability and Environmental Compliance: The process is inherently designed for commercial scale-up, with reaction conditions that are easily transferable from pilot plants to multi-ton reactors. The reduction in hazardous waste generation, particularly through the minimized use of strong oxidants and the efficient recycling of solvents, aligns with modern environmental, social, and governance (ESG) goals. This compliance facilitates smoother regulatory approvals and strengthens the manufacturer's reputation as a sustainable partner in the fine chemical industry.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production of 6 alpha, 9 alpha-diflupredone, derived directly from the patent specifications and process capabilities. These insights are intended to clarify the operational benefits and quality assurances associated with this advanced synthetic method. Understanding these details is crucial for stakeholders evaluating the feasibility of integrating this intermediate into their broader drug development pipelines.
Q: Why is fluoroboric acid preferred over perchloric acid in this synthesis?
A: Fluoroboric acid is used instead of perchloric acid to eliminate the risk of combustion and explosion associated with strong oxidants, significantly enhancing production safety while reducing oxidation impurities.
Q: How does this route improve impurity profiles compared to conventional methods?
A: By utilizing anecortave dehydroacetate as the starting material and employing a one-pot fluorination strategy, the process minimizes isomer formation and avoids the hydrolysis of ester groups that typically occurs in alkaline epoxy formation steps of older routes.
Q: What are the scalability advantages of this preparation method?
A: The method features a short reaction route with high overall yield (>54%) and high purity (>98%), using readily available raw materials and safer reagents, making it highly suitable for commercial scale-up from kilograms to metric tons.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 6 Alpha 9 Alpha Diflupredone Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of securing a reliable 6 alpha, 9 alpha-diflupredone supplier who can deliver both technical excellence and commercial reliability. As a premier CDMO expert, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project needs are met with precision and speed. Our facilities are equipped with rigorous QC labs and adhere to stringent purity specifications, guaranteeing that every batch of hormone intermediate meets the highest international standards. We understand that the transition from patent to production requires a partner who can navigate complex regulatory landscapes while maintaining cost efficiency, and our team is dedicated to providing that seamless support.
We invite you to collaborate with us to unlock the full potential of this innovative synthesis route for your pharmaceutical applications. Please contact our technical procurement team to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our manufacturing capabilities can enhance your supply chain stability and drive down your overall production costs.