Industrial Scale Synthesis of Aryl-iminomethyl-carbamates for High-Purity Pharmaceutical Intermediates
The pharmaceutical industry continuously demands robust synthetic routes for high-value intermediates, particularly those exhibiting potent biological activity such as LTB4 antagonists. Patent CN1541999A introduces a groundbreaking industrial process for the preparation of aryl-iminomethyl-carbamates, specifically targeting compounds of general formula (I) with exceptional purity profiles. This methodology addresses the critical need for scalable synthesis in the production of advanced pharmaceutical intermediates, leveraging specific silylation strategies to overcome traditional yield limitations. By utilizing alkali metal hexaalkyldisilazane reagents, the process ensures a highly controlled transformation of nitrile precursors into the desired carbamate structures. The strategic implementation of this technology enables manufacturers to achieve consistent quality standards required for regulatory compliance in global markets. Furthermore, the isolation of key intermediates as crystalline hydrochloride salts provides a significant advantage in purification efficiency, reducing the burden on downstream processing units. This technical advancement represents a pivotal shift towards more reliable and cost-effective manufacturing paradigms for complex organic molecules used in therapeutic applications.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthetic pathways for generating aryl-iminomethyl-carbamates often suffer from significant inefficiencies that hinder their viability for large-scale commercial operations. Conventional methods frequently rely on harsh reaction conditions that can lead to the degradation of sensitive functional groups, resulting in complex impurity profiles that are difficult to resolve. The lack of selective crystallization steps in older processes means that extensive chromatographic purification is often required, which drastically increases production costs and extends lead times for high-purity pharmaceutical intermediates. Additionally, the use of unstable intermediates in legacy routes poses safety risks and complicates the supply chain continuity for critical raw materials. Many existing protocols fail to provide a robust mechanism for removing trace metal contaminants or organic by-products, which is unacceptable for modern drug substance manufacturing. The inability to consistently achieve high yields without compromising purity remains a persistent bottleneck in the industry. These limitations necessitate a re-evaluation of synthetic strategies to ensure economic feasibility and operational safety in contemporary chemical manufacturing environments.
The Novel Approach
The innovative process described in the patent data revolutionizes the synthesis landscape by introducing a highly selective silylation and hydrolysis sequence that maximizes yield while minimizing impurity formation. By employing alkali metal hexaalkyldisilazane, specifically lithium hexamethyldisilazane, the reaction achieves superior nucleophilic activation of the nitrile group under mild thermal conditions. This approach facilitates a clean conversion to the intermediate amidine species without the generation of excessive side products that typically plague conventional carbamate synthesis. A defining feature of this novel route is the strategic isolation of the intermediate as a hydrochloride salt, which crystallizes readily from the reaction mixture, allowing for the physical exclusion of soluble impurities. The use of common industrial solvents such as tetrahydrofuran and toluene further enhances the practicality of this method for commercial scale-up of complex pharmaceutical intermediates. The process design inherently supports cost reduction in pharmaceutical intermediates manufacturing by simplifying work-up procedures and reducing solvent consumption. This methodological shift ensures that the final product meets stringent purity specifications with minimal additional processing effort.
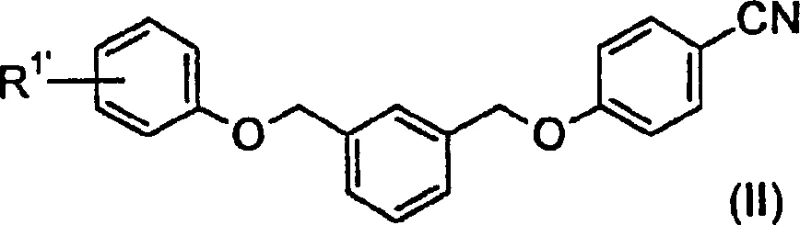
Mechanistic Insights into Hexaalkyldisilazane Mediated Nitrile Conversion
The core chemical transformation relies on the precise deprotonation and silylation of the nitrile functionality within the Formula (II) substrate using a strong non-nucleophilic base. When the nitrile compound is treated with lithium hexamethyldisilazane in an ether solvent like tetrahydrofuran, a silylated ketenimine intermediate is generated in situ. This reactive species is then captured by an electrophilic carbamoyl source, such as ethyl chloroformate, to form the protected carbamate structure. The reaction kinetics are carefully managed by maintaining temperatures between -50°C and 30°C during the addition phase to prevent thermal decomposition of the sensitive silylated intermediate. Subsequent hydrolysis with aqueous hydrochloric acid cleaves the silyl protecting groups and protonates the amidine nitrogen, driving the formation of the stable hydrochloride salt. This mechanistic pathway is distinct from direct amidination methods as it avoids the use of toxic ammonia sources or high-pressure conditions. The controlled addition of reagents ensures that the reaction proceeds with high atom economy, reducing the generation of hazardous waste streams. Understanding this mechanism is crucial for R&D teams aiming to replicate the high purity levels reported in the patent documentation for their own process development initiatives.
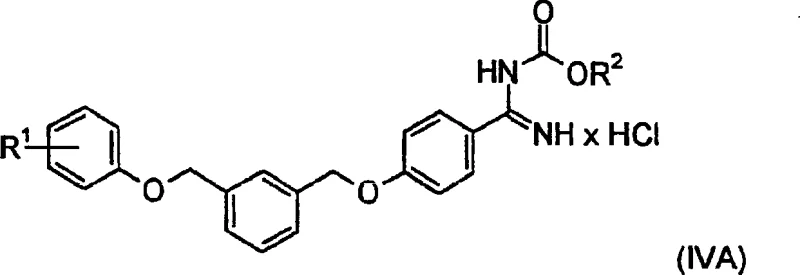
Impurity control is intrinsically built into the chemical design of this synthesis through the formation of the crystalline hydrochloride salt of Formula (IVA). The salt formation step acts as a powerful purification tool, as the specific crystal lattice structure of the hydrochloride excludes structurally similar by-products and unreacted starting materials. By adjusting the solvent composition during crystallization, using mixtures of acetone and methyl isobutyl ketone, operators can fine-tune the solubility parameters to maximize the recovery of the desired product. The use of dilute hydrochloric acid for hydrolysis ensures that the pH is maintained at a level that favors salt precipitation without causing acid-catalyzed degradation of the carbamate linkage. This dual function of reaction and purification significantly reduces the need for additional chromatographic steps, which are often the most costly part of intermediate production. The resulting solid can be isolated via centrifugation and washed with organic solvents to remove residual mother liquor contaminants. This robust impurity rejection mechanism guarantees that the final free base, obtained after neutralization, possesses the high-purity profile required for downstream pharmaceutical applications.
How to Synthesize Aryl-iminomethyl-carbamates Efficiently
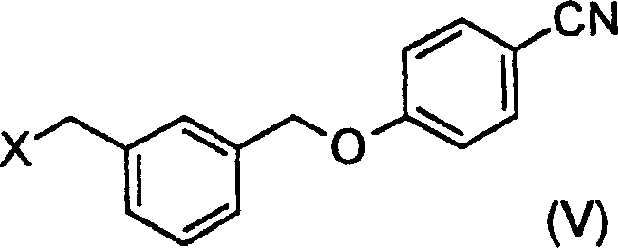
The synthesis of these high-value compounds requires a disciplined approach to reaction parameter control and intermediate handling to ensure optimal outcomes. The process begins with the preparation of the silylated nitrile intermediate, followed by electrophilic trapping and acidic work-up to isolate the stable salt form. Detailed operational guidelines regarding temperature ramps, addition rates, and solvent ratios are essential for reproducing the high yields observed in the patent examples. Operators must pay close attention to the moisture content of the solvents, as the presence of water can prematurely quench the silylating agent and reduce overall conversion efficiency. The neutralization step to release the free base must be performed under buffered conditions to prevent localized high pH zones that could degrade the product. Adhering to these standardized protocols ensures that the commercial scale-up of complex pharmaceutical intermediates proceeds smoothly without unexpected deviations. For a comprehensive breakdown of the specific operational parameters and equipment requirements, please refer to the standardized synthesis steps provided below.
- React Formula (II) nitrile with lithium hexamethyldisilazane in THF at controlled low temperatures to form the silylated intermediate.
- Treat the reaction mixture with ethyl chloroformate followed by hydrolysis with aqueous hydrochloric acid to isolate the crystalline hydrochloride salt.
- Neutralize the hydrochloride salt using trisodium citrate dihydrate in an organic solvent system to release the high-purity free base.
Commercial Advantages for Procurement and Supply Chain Teams
This patented synthesis route offers profound benefits for procurement and supply chain management by fundamentally altering the cost structure of producing these specialized intermediates. The ability to isolate a crystalline intermediate salt significantly reduces the complexity of the purification train, leading to substantial cost savings in manufacturing overhead and utility consumption. By eliminating the need for extensive chromatographic purification, the process shortens the production cycle time, thereby enhancing supply chain reliability and reducing lead time for high-purity pharmaceutical intermediates. The use of commercially available reagents like lithium hexamethyldisilazane and ethyl chloroformate ensures that raw material sourcing remains stable and unaffected by niche supply constraints. Furthermore, the robustness of the crystallization step provides a consistent quality output that minimizes the risk of batch rejection and rework. These factors collectively contribute to a more resilient supply chain capable of meeting the rigorous demands of global pharmaceutical clients. The process design inherently supports scalability, allowing for seamless transition from pilot plant to full commercial production volumes.
- Cost Reduction in Manufacturing: The elimination of expensive transition metal catalysts and the reduction in solvent usage for purification directly translate to lower variable costs per kilogram of product. By utilizing a crystallization-driven purification strategy, the process avoids the high operational expenses associated with preparative HPLC or flash chromatography columns. The high yield of the hydrochloride salt isolation step ensures that raw material costs are amortized over a larger quantity of saleable product, improving overall margin potential. Additionally, the recycling of mother liquors is facilitated by the simplicity of the solvent system, further driving down waste disposal costs. This economic efficiency makes the process highly attractive for cost-sensitive procurement strategies in the competitive fine chemical market.
- Enhanced Supply Chain Reliability: The reliance on stable, shelf-stable reagents and standard industrial solvents mitigates the risk of supply disruptions caused by specialized chemical shortages. The robust nature of the reaction conditions allows for flexible scheduling and batch processing, ensuring consistent delivery timelines to downstream customers. The ability to store the intermediate hydrochloride salt for extended periods without degradation provides a strategic buffer against demand fluctuations. This stability enhances the overall reliability of the supply chain, allowing procurement managers to plan inventory levels with greater confidence. The process is designed to be resilient against minor variations in raw material quality, further securing the continuity of supply for critical pharmaceutical projects.
- Scalability and Environmental Compliance: The process is inherently scalable, as demonstrated by the patent examples which utilize kilogram-scale quantities without loss of efficiency. The use of aqueous work-ups and common organic solvents simplifies waste treatment and aligns with modern environmental compliance standards. The reduction in hazardous waste generation through high-yield crystallization supports corporate sustainability goals and reduces regulatory burden. The equipment requirements are standard for fine chemical manufacturing, facilitating easy technology transfer between production sites. This scalability ensures that the supply can grow in tandem with market demand for the final therapeutic agent without requiring significant capital investment in new specialized infrastructure.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These answers are derived directly from the detailed specifications and examples provided in the patent documentation to ensure accuracy. Understanding these aspects is vital for technical teams evaluating the feasibility of adopting this route for their specific production needs. The information covers critical parameters such as reagent selection, safety protocols, and quality control measures. This section aims to provide clarity on the operational advantages and technical requirements associated with the process.
Q: How does the hydrochloride salt formation improve purity?
A: The formation of the well-crystalline hydrochloride salt (Formula IVA) allows for the effective removal of by-products and impurities through crystallization, significantly enhancing the purity profile of the final pharmaceutical intermediate.
Q: What are the key safety considerations for using LiHMDS?
A: Lithium hexamethyldisilazane is moisture-sensitive and requires handling under inert atmosphere conditions. The process mandates strict temperature control between -50°C and 30°C during addition to prevent exothermic runaway.
Q: Is this process suitable for large-scale commercial production?
A: Yes, the patent explicitly describes industrial-scale examples using kilogram quantities of reagents, demonstrating the feasibility of commercial scale-up for complex pharmaceutical intermediates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Aryl-iminomethyl-carbamates Supplier
The technical potential of this synthesis route is immense, offering a pathway to high-quality intermediates that meet the rigorous standards of the global pharmaceutical industry. NINGBO INNO PHARMCHEM, as a seasoned CDMO expert, possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our facility is equipped with stringent purity specifications and rigorous QC labs to ensure that every batch of aryl-iminomethyl-carbamates delivered meets the highest quality benchmarks. We understand the critical nature of these intermediates in the drug development timeline and are committed to providing seamless support from process optimization to commercial supply. Our team of chemists is ready to adapt this patented methodology to fit specific client requirements while maintaining full regulatory compliance.
We invite you to initiate a conversation about optimizing your supply chain for these critical compounds. Our technical procurement team is prepared to provide a Customized Cost-Saving Analysis tailored to your specific volume needs. We encourage potential partners to contact us to request specific COA data and route feasibility assessments for your projects. By collaborating with us, you gain access to a reliable partner dedicated to driving efficiency and quality in your pharmaceutical manufacturing operations. Let us help you secure a stable and cost-effective supply of these essential building blocks for your next generation of therapies.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →