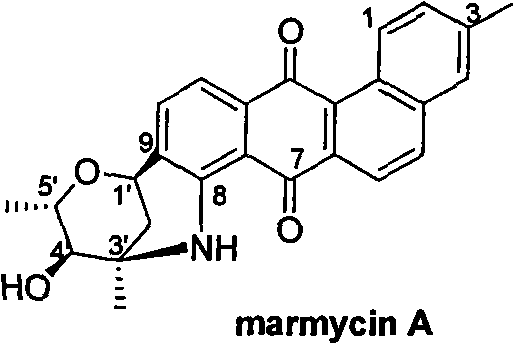
Advanced Synthetic Route for Novel Benzanthracycline Anticancer Intermediates
Introduction to Patent CN101993446B Technology
The pharmaceutical industry continuously seeks robust synthetic pathways for complex natural product derivatives that exhibit potent biological activity. Patent CN101993446B introduces a groundbreaking methodology for the synthesis of novel benzanthracycline compounds, specifically focusing on 3'-demethylated derivatives of the marine natural product Marmycin A. These compounds are structurally classified as polyketide-derived angular tetracyclic anthraquinones, distinguished by a unique hexacyclic skeleton featuring simultaneous carbon-carbon and carbon-nitrogen glycosidic linkages. Unlike traditional anthracyclines that rely on single glycosidic bonds, this architecture incorporates four chiral centers and a challenging quaternary carbon, presenting significant synthetic hurdles that this patent successfully overcomes. The technology provides a reliable pharmaceutical intermediate supplier with the capability to produce high-purity analogues for extensive pharmacological evaluation.
The core innovation lies in the ability to bypass the limitations of natural extraction, which often yields insufficient quantities for comprehensive drug discovery programs. By establishing a concise, metal-catalyzed synthetic route, the patent enables the rapid generation of diverse libraries of benzanthracycline derivatives. This approach not only secures a stable supply chain for research materials but also opens avenues for structure-activity relationship (SAR) studies that were previously impossible due to the scarcity of the natural product. The compounds have demonstrated significant potential as topoisomerase inhibitors, showing strong cytotoxicity against human colon cancer cells, thereby validating the commercial and scientific value of this synthetic methodology for developing next-generation anticancer agents.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, accessing Marmycin A and its analogues has been plagued by severe inefficiencies inherent to natural product isolation. The natural source, marine actinomycetes, offers extremely low titers, making large-scale fermentation economically unviable for industrial applications. Furthermore, the complex molecular architecture of Marmycin A, particularly the presence of a quaternary carbon center and the fused hexacyclic system, renders total synthesis exceptionally difficult. Prior to this invention, no reports existed regarding the total synthesis of Marmycin A, leaving researchers dependent on trace amounts isolated from nature. This dependency creates a bottleneck in drug discovery, as the inability to rapidly derivatize the sugar moiety prevents the optimization of pharmacokinetic properties and toxicity profiles. Conventional glycosylation strategies often struggle to control stereochemistry at multiple centers simultaneously, leading to complex mixtures that are costly and time-consuming to separate.
The Novel Approach
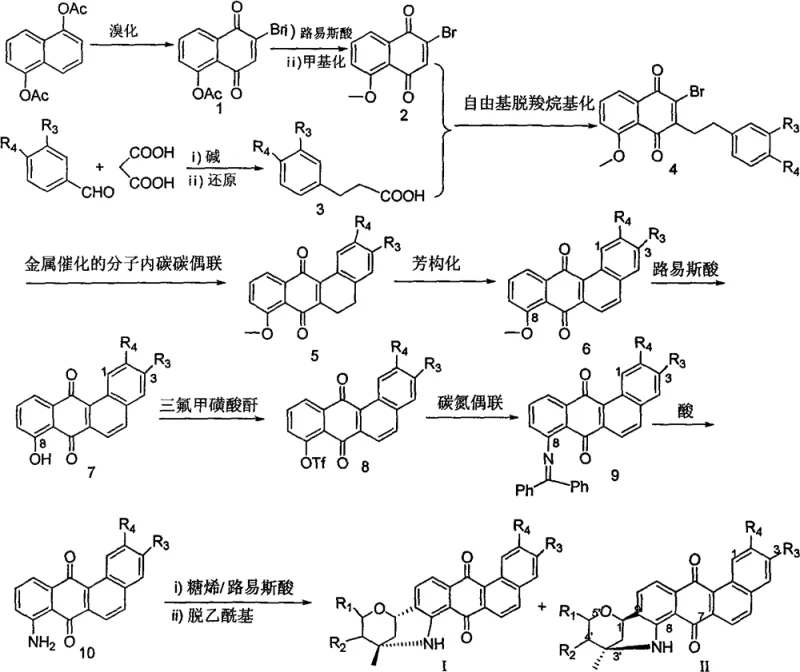
The methodology disclosed in the patent represents a paradigm shift by employing a highly regioselective, metal-catalyzed strategy to construct the core benzanthraquinone skeleton efficiently. Instead of struggling with the natural product's complexity directly, the process builds the aglycone through a streamlined sequence involving intramolecular carbon-carbon coupling. This step is critical as it establishes the angular tetracyclic framework with high precision, avoiding the formation of unwanted regioisomers that typically plague such cyclizations. Subsequently, the transformation of the 8-methoxy group to an amino group via metal-catalyzed carbon-nitrogen coupling provides a versatile handle for the final glycosylation. This modular approach allows for the interchangeability of sugar components, enabling the synthesis of a wide array of derivatives without redesigning the entire synthetic route, thus offering substantial cost savings in manufacturing complex pharmaceutical intermediates.
Mechanistic Insights into Lewis Acid-Catalyzed Glycosylation
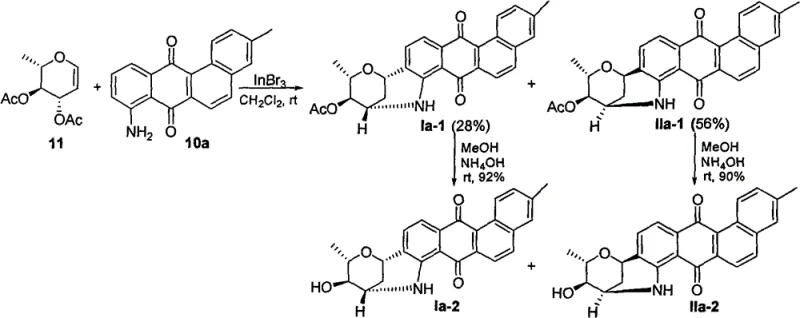
The crown jewel of this synthetic strategy is the final glycosylation step, which elegantly solves the problem of constructing the unique C-C and C-N linked hexacyclic system. Under the catalysis of Lewis acids such as Indium(III) bromide (InBr3), the 8-aminobenzanthraquinone intermediate reacts with acetylated sugar alkenes. This reaction is not a simple substitution but a concerted process where the nucleophilic amino group and the electron-rich aromatic ring simultaneously attack the activated sugar alkene. This dual attack results in the formation of both a carbon-nitrogen bond and a carbon-carbon bond in a single operation, locking the sugar moiety into the rigid hexacyclic framework. The mechanism ensures high stereocontrol, yielding specific epimers (Formula I with S,S configuration and Formula II with R,R configuration at C1' and C3') depending on the reaction conditions and substrate geometry. This level of control is paramount for maintaining the biological activity associated with the specific three-dimensional arrangement of the pharmacophore.
Furthermore, the choice of Lewis acid catalyst is instrumental in managing the impurity profile of the final product. Traditional acidic conditions might lead to decomposition of the sensitive anthraquinone core or hydrolysis of the acetyl protecting groups prematurely. However, the mild yet effective Lewis acid conditions specified in the patent promote the desired cyclization while preserving the integrity of the protecting groups until the final deacetylation step. This sequential protection-deprotection strategy simplifies purification, as the acetylated intermediates can be easily separated via silica gel chromatography before the final global deprotection. The result is a high-purity product suitable for biological testing, minimizing the risk of false positives in assays caused by synthetic impurities, which is a critical consideration for any reliable pharmaceutical intermediate supplier aiming to support rigorous drug discovery pipelines.
How to Synthesize 3'-Demethylated Marmycin A Derivatives Efficiently
The synthesis protocol outlined in the patent is designed for operational simplicity and scalability, making it ideal for translation from laboratory bench to pilot plant production. The process begins with readily available starting materials like 1,5-diacetoxynaphthalene and substituted benzaldehydes, progressing through nine distinct chemical transformations. Each step has been optimized for yield and purity, utilizing standard organic synthesis techniques such as reflux, extraction, and column chromatography. The detailed procedure ensures that the critical stereocenters are established correctly during the glycosylation phase, followed by a straightforward deacetylation using aqueous ammonia or carbonate bases. For a comprehensive understanding of the specific reaction parameters, solvent systems, and workup procedures required to replicate these results, please refer to the standardized synthesis guide below.
- Construct the 8-methoxybenzanthraquinone skeleton via metal-catalyzed intramolecular carbon-carbon coupling with high regioselectivity.
- Convert the 8-methoxy group to an amino group through efficient metal-catalyzed carbon-nitrogen coupling to obtain the key 8-aminobenzanthraquinone intermediate.
- Perform Lewis acid-catalyzed glycosylation with acetylated sugar alkenes to simultaneously form carbon-carbon and carbon-nitrogen glycosidic bonds, followed by deacetylation.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthetic route offers transformative advantages over relying on natural extraction or legacy synthetic methods. The primary benefit is the drastic simplification of the supply chain; by synthesizing the core scaffold from commodity chemicals, manufacturers are no longer vulnerable to the fluctuations and geopolitical risks associated with sourcing rare marine organisms. This independence ensures a continuous and reliable supply of high-purity intermediates, which is essential for maintaining uninterrupted clinical trial timelines. Moreover, the high regioselectivity of the metal-catalyzed coupling steps minimizes the generation of difficult-to-remove isomers, significantly reducing the burden on downstream purification processes and lowering the overall cost of goods sold (COGS).
- Cost Reduction in Manufacturing: The synthetic route eliminates the need for expensive and low-yield fermentation processes. By utilizing efficient catalytic cycles with recoverable metals like palladium and indium, the process maximizes atom economy. The high yields reported in key steps, such as the demethylation and triflation reactions, mean less raw material waste and lower solvent consumption per kilogram of product. Additionally, the ability to produce diverse analogues from a common intermediate allows for economies of scale, further driving down the unit cost of these specialized anticancer intermediates without compromising on quality standards.
- Enhanced Supply Chain Reliability: Dependence on natural sources introduces significant variability in batch-to-batch consistency and availability. This synthetic method mitigates those risks by establishing a defined chemical manufacturing process that can be validated and scaled. The use of robust reaction conditions that tolerate a range of functional groups ensures that the supply of critical intermediates remains stable even if specific sugar precursors need to be swapped. This flexibility allows procurement teams to secure long-term contracts with confidence, knowing that the manufacturing partner can adapt to changing project requirements without disrupting the flow of materials.
- Scalability and Environmental Compliance: The process is inherently scalable, having been demonstrated effectively on gram scales in the patent examples with clear pathways to kilogram and tonne production. The reaction conditions avoid the use of extremely hazardous reagents or cryogenic temperatures, facilitating safer operations in large-scale reactors. Furthermore, the streamlined number of steps reduces the total volume of chemical waste generated. The ability to recycle solvents and the high efficiency of the catalytic steps contribute to a greener manufacturing footprint, aligning with increasingly stringent environmental regulations and corporate sustainability goals in the fine chemical sector.
Frequently Asked Questions (FAQ)
Understanding the technical nuances of this synthetic technology is crucial for stakeholders evaluating its potential for their drug development programs. The following questions address common inquiries regarding the scalability, mechanism, and application of these benzanthracycline derivatives. The answers are derived directly from the experimental data and technical specifications provided in the patent documentation, ensuring accuracy and relevance for technical decision-makers.
Q: What is the primary advantage of this synthetic method over natural extraction?
A: Natural Marmycin A is difficult to extract in large quantities and possesses a complex quaternary carbon center that hinders derivatization. This synthetic route provides a scalable, high-yield alternative that allows for structural modification at the 3'-position for optimized pharmacological screening.
Q: How does the glycosylation step achieve the unique hexacyclic structure?
A: The process utilizes a Lewis acid catalyst to facilitate a tandem reaction where both carbon-carbon and carbon-nitrogen glycosidic bonds are formed simultaneously between the 8-aminobenzanthraquinone and the sugar alkene, creating the characteristic hexacyclic skeleton with four chiral centers.
Q: What are the biological applications of these benzanthracycline compounds?
A: These compounds function as potent topoisomerase TopI inhibitors. They exhibit significant inhibitory activity against various cancer cell lines, including human colon cancer (HCT-116), leukemia (HL-60), and lung cancer (A-549), making them valuable candidates for anticancer drug development.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Benzanthracycline Compounds Supplier
As the demand for sophisticated anticancer intermediates grows, partnering with an experienced CDMO is essential for translating patented chemistry into commercial reality. NINGBO INNO PHARMCHEM possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can move seamlessly from preclinical research to late-stage clinical supply. Our facility is equipped with state-of-the-art rigorous QC labs capable of meeting stringent purity specifications required for pharmaceutical applications. We understand the critical nature of topoisomerase inhibitors and the need for precise stereochemical control, and our team is dedicated to delivering materials that consistently meet these high standards.
We invite you to collaborate with us to leverage this advanced synthetic technology for your oncology pipeline. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and timeline. Contact us today to request specific COA data for our benzanthracycline intermediates and discuss route feasibility assessments for your target molecules. Let us help you accelerate your drug discovery efforts with reliable, high-quality chemical solutions.