Advancing C-Aryl Glycoside Production with Novel Palladium Catalysis for Commercial Scale-Up
The pharmaceutical industry continuously seeks robust synthetic routes for complex carbohydrate derivatives, particularly C-aryl glycosides, which serve as critical scaffolds in modern drug design. Patent CN111269205B introduces a groundbreaking preparation method that addresses long-standing challenges in constructing stable C-glycosidic bonds. This technology leverages a sophisticated palladium-catalyzed cross-coupling strategy to link halogenated aromatic hydrocarbons with glycosyl chlorides efficiently. Unlike traditional approaches that often struggle with stereocontrol, this novel protocol ensures high stereoselectivity and operational simplicity. For R&D directors and procurement specialists, understanding this methodology is vital as it unlocks access to high-purity intermediates essential for developing next-generation antidiabetic and anticancer therapeutics. The process operates under relatively mild thermal conditions, typically ranging from 80°C to 120°C, which significantly reduces energy consumption and safety risks associated with high-temperature operations.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of C-aryl glycosidic bonds has relied heavily on two primary strategies, both of which present significant drawbacks for large-scale manufacturing. The first approach involves Friedel-Crafts type glycosylation using equivalent Lewis acids to catalyze the reaction between electron-rich aromatics and glycosyl donors. This method frequently suffers from poor regioselectivity and stereoselectivity, leading to complex mixtures that are difficult and costly to separate. Furthermore, the use of stoichiometric amounts of Lewis acids generates substantial waste, complicating environmental compliance and increasing disposal costs. The second conventional route utilizes pre-functionalized aryl metal reagents coupled with glycosyl donors. While effective in some contexts, the requirement for pre-functionalization adds multiple synthetic steps, reducing overall atom economy and increasing lead times. These limitations hinder the rapid development of diverse compound libraries needed for drug discovery programs.
The Novel Approach
The methodology disclosed in CN111269205B represents a paradigm shift by employing a direct palladium-catalyzed coupling system that bypasses the need for harsh Lewis acids or pre-functionalized organometallic species. By utilizing a specific combination of a palladium catalyst, a specialized ligand, and a unique co-catalyst, the reaction achieves excellent stereocontrol directly from glycosyl chlorides. This approach not only simplifies the synthetic sequence but also expands the substrate scope to include a wide variety of halogenated aromatics and termination reagents such as alkenes, alcohols, and alkynes. The ability to synthesize C-aryl glycoside compounds that are difficult to obtain by other methods provides a distinct competitive advantage. Moreover, the post-treatment process is streamlined, involving simple extraction and chromatography, which facilitates easier purification and higher final purity levels suitable for pharmaceutical applications.
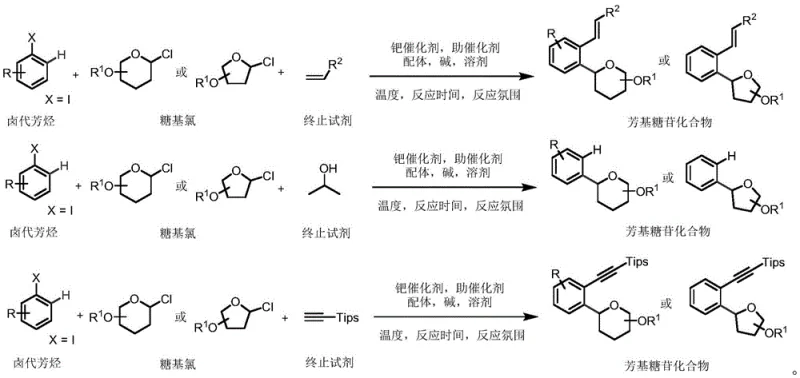
Mechanistic Insights into Pd-Catalyzed C-Glycosylation
The success of this synthetic route lies in the intricate interplay between the palladium catalyst and the specific co-catalyst system. The reaction initiates with the oxidative addition of the halogenated aromatic hydrocarbon to the palladium center, forming an aryl-palladium species. The presence of the co-catalyst, structurally defined as a bicyclic amide derivative, plays a crucial role in stabilizing the active catalytic species and facilitating the subsequent transmetallation or insertion steps. This co-catalyst helps to modulate the electronic environment around the metal center, ensuring that the glycosyl chloride reacts with high fidelity. The ligand, such as tri(2-furyl)phosphine, further enhances the stability and reactivity of the catalyst, preventing premature decomposition and promoting the desired coupling pathway over side reactions.
Impurity control is a paramount concern in the synthesis of pharmaceutical intermediates, and this method excels in minimizing byproduct formation. The high stereoselectivity observed is attributed to the rigid coordination geometry enforced by the catalyst-ligand-co-catalyst complex, which directs the approach of the glycosyl donor. This minimizes the formation of anomeric mixtures, a common issue in glycosylation chemistry. Additionally, the use of mild bases like cesium carbonate or potassium carbonate prevents the degradation of sensitive functional groups on the aromatic ring or the sugar moiety. By avoiding strong acids or bases, the process preserves the integrity of protecting groups, reducing the need for re-protection steps and thereby improving overall yield. This level of control is essential for producing high-purity API intermediates that meet stringent regulatory standards.
How to Synthesize C-Aryl Glycoside Efficiently
Implementing this synthesis requires careful attention to reaction parameters to maximize efficiency and yield. The process begins by charging a reaction vessel with the necessary reagents under an inert nitrogen atmosphere to prevent catalyst oxidation. The molar ratios are critical, with the base typically used in excess relative to the halogenated aromatic hydrocarbon to drive the reaction to completion. Solvent selection is also flexible, with polar aprotic solvents like tetrahydrofuran or acetonitrile proving effective. Detailed standardized synthesis steps follow below to guide process chemists in replicating these results reliably.
- Combine base, co-catalyst, palladium catalyst, ligand, halogenated aromatic hydrocarbon, glycosyl chloride, termination reagent, and organic solvent in a nitrogen-purged vessel.
- React the mixture at 80-120°C for 15-30 hours to obtain the crude material.
- Dilute with ethyl acetate, wash with water, separate the organic phase, dry, filter, concentrate, and purify via chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this technology offers tangible benefits beyond mere technical feasibility. The streamlined nature of the reaction sequence translates directly into reduced manufacturing complexity. By eliminating the need for pre-functionalized aryl metal reagents, the number of unit operations is significantly decreased, which lowers capital expenditure requirements for equipment and reduces the footprint of the production facility. The use of commercially available starting materials, such as simple halogenated aromatics and glycosyl chlorides, ensures a stable and reliable supply chain, mitigating the risk of raw material shortages that can plague more exotic synthetic routes.
- Cost Reduction in Manufacturing: The elimination of stoichiometric Lewis acids and the reduction in synthetic steps lead to substantial cost savings in raw material consumption and waste disposal. The simplified workup procedure, which avoids complex quenching protocols associated with reactive organometallics, further reduces labor and utility costs. Additionally, the high yields reported in the patent examples indicate efficient resource utilization, maximizing the output per batch and lowering the cost of goods sold for the final intermediate.
- Enhanced Supply Chain Reliability: The robustness of the catalyst system allows for consistent performance across different batches, ensuring predictable delivery schedules. The tolerance of the reaction to various functional groups means that a single platform technology can be used to produce a diverse range of derivatives, simplifying inventory management. This flexibility allows manufacturers to respond quickly to changing market demands without needing to retool or develop entirely new processes for each new compound variant.
- Scalability and Environmental Compliance: The reaction conditions are mild enough to be safely scaled from laboratory to pilot and commercial production scales without significant engineering challenges. The use of standard organic solvents and the generation of less hazardous waste align with green chemistry principles, facilitating easier regulatory approval and reducing the environmental impact of manufacturing operations. This compliance is increasingly important for maintaining social license to operate and meeting corporate sustainability goals.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this C-aryl glycoside synthesis method. These answers are derived directly from the experimental data and claims within the patent documentation, providing a reliable basis for decision-making. Understanding these details helps stakeholders assess the feasibility of integrating this technology into their existing production workflows.
Q: What are the key advantages of this Pd-catalyzed method over traditional Friedel-Crafts glycosylation?
A: This method offers superior regioselectivity and stereoselectivity compared to traditional Lewis acid-catalyzed Friedel-Crafts reactions, which often suffer from poor control and harsh conditions. Additionally, it avoids the cumbersome pre-functionalization required for aryl metal reagents.
Q: What is the typical reaction temperature and time for this synthesis?
A: The optimal reaction conditions involve heating the mixture to between 90°C and 110°C for approximately 24 hours, ensuring high conversion while maintaining the stability of sensitive glycosyl donors.
Q: Can this method be scaled for industrial production of SGLT2 inhibitor intermediates?
A: Yes, the process utilizes readily available starting materials and standard organic solvents like tetrahydrofuran or acetonitrile, making it highly suitable for commercial scale-up with simplified post-treatment procedures.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable C-Aryl Glycoside Supplier
NINGBO INNO PHARMCHEM stands at the forefront of adopting advanced synthetic methodologies to deliver high-quality pharmaceutical intermediates. Our expertise in scaling diverse pathways from 100 kgs to 100 MT/annual commercial production ensures that we can meet the rigorous demands of global supply chains. We possess extensive experience in handling complex catalytic systems, including the palladium-catalyzed processes described herein, guaranteeing stringent purity specifications through our rigorous QC labs. Our commitment to quality ensures that every batch of C-aryl glycoside intermediate meets the highest industry standards.
We invite potential partners to engage with our technical procurement team to discuss how this innovative synthesis route can optimize your supply chain. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into the economic benefits of switching to this method. We encourage you to contact us for specific COA data and route feasibility assessments tailored to your project needs, ensuring a seamless transition to more efficient and cost-effective manufacturing solutions.