Advanced Synthesis of Moxifloxacin Degradation Impurity J for Global Quality Control Standards
Advanced Synthesis of Moxifloxacin Degradation Impurity J for Global Quality Control Standards
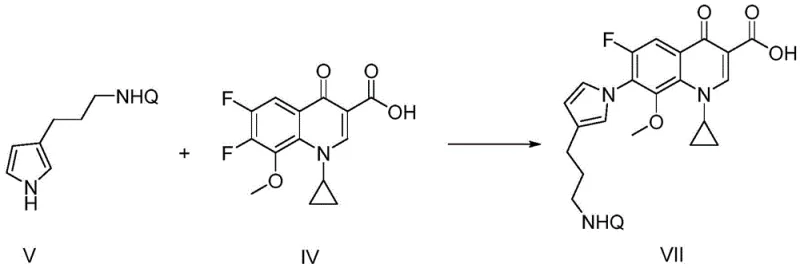
The pharmaceutical industry relies heavily on the availability of high-purity reference standards to ensure the safety and efficacy of active pharmaceutical ingredients (APIs). A critical development in this domain is disclosed in patent CN110627768B, which details a robust and efficient preparation method for Moxifloxacin degradation impurity J. Moxifloxacin hydrochloride, a fourth-generation fluoroquinolone antibacterial agent, is known to undergo photodegradation during stability testing, generating specific impurities that must be strictly monitored. Historically, obtaining these degradation products in sufficient quantity and purity for analytical validation has been a significant bottleneck, often relying on difficult isolation procedures from degraded drug substances. This new patented methodology revolutionizes the supply chain for this critical quality control material by establishing a total synthesis route that begins with pyrrole-3-formaldehyde. By shifting from isolation to synthesis, the process guarantees a consistent, scalable, and high-yield source of Impurity J, addressing a vital need for regulatory compliance and quality assurance in the global fluoroquinolone market.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior to the innovation described in patent CN110627768B, the acquisition of Moxifloxacin degradation impurity J was fraught with technical and economic inefficiencies. The existing literature, such as reports in AAPS PharmSciTech, primarily describes isolation methods where the impurity is extracted from Moxifloxacin hydrochloride that has been subjected to forced photodegradation. This approach is inherently flawed for several reasons. Firstly, the concentration of the degradation impurity in the photolyzed mixture is typically extremely low, making the isolation process labor-intensive and resulting in dismal overall yields. Secondly, the separation of the target impurity from the complex matrix of other degradation products and the remaining API requires sophisticated and costly chromatographic techniques, which are difficult to scale beyond milligram quantities. Furthermore, the purity of the isolated material is often compromised by co-eluting degradation byproducts, rendering it less suitable as a precise reference standard for high-performance liquid chromatography (HPLC) quantification. These limitations create a fragile supply chain where the availability of the reference standard is tied to the wasteful degradation of valuable finished drug product.
The Novel Approach
The novel approach presented in the patent data fundamentally alters the production landscape by employing a convergent synthetic strategy that constructs the impurity molecule from simple, commercially available building blocks. Instead of degrading the final drug, the inventors utilize pyrrole-3-formaldehyde as a raw material to construct the specific side chain required for Impurity J. This side chain is synthesized through a sequence of Wittig reaction, reduction, and amino protection steps, allowing for precise control over the molecular architecture before it is ever coupled to the quinoline core. The subsequent nucleophilic substitution with 1-cyclopropyl-6,7-difluoro-8-methoxy-1,4-dihydro-4-oxo-3-quinolinecarboxylic acid is optimized to maximize yield and minimize byproduct formation. Finally, a controlled deprotection step releases the free amine to yield the target Impurity J. This synthetic route not only achieves a purity exceeding 99% but also offers substantially higher yields compared to isolation. By decoupling the production of the reference standard from the degradation of the API, this method ensures a stable, independent, and economically viable supply source for pharmaceutical quality control laboratories worldwide.
Mechanistic Insights into the Multi-Step Synthetic Route
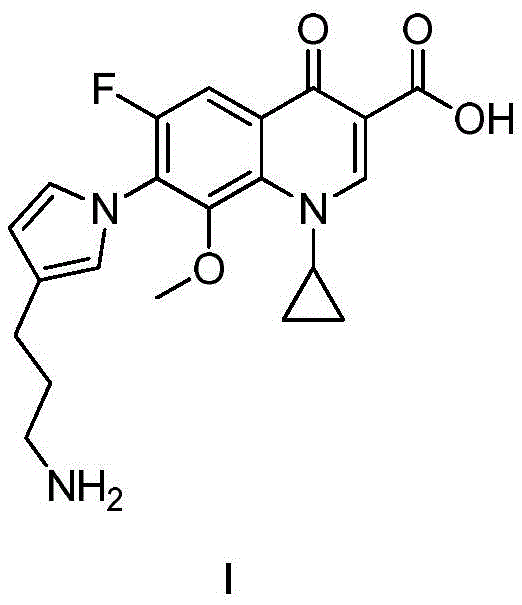
The chemical elegance of this synthesis lies in its stepwise construction of the pyrrole-propylamine side chain and its subsequent attachment to the fluoroquinolone nucleus. The process initiates with a Wittig reaction, where pyrrole-3-formaldehyde reacts with a triphenyl acetonitrile-based quaternary phosphonium salt under alkaline conditions. This transformation extends the carbon chain by two carbons and introduces a nitrile group, forming pyrrole-3-acrylonitrile. The choice of base and solvent in this step is critical; the patent specifies the use of bases like sodium hydride or DBU in solvents such as toluene or THF to drive the equilibrium towards the alkene product while minimizing polymerization of the reactive aldehyde. Following this, a crucial reduction phase converts both the olefinic double bond and the nitrile triple bond into a saturated propylamine chain. This can be achieved via a one-pot high-pressure hydrogenation or a stepwise approach using catalysts like Pd/C or reducing agents like LiAlH4. The versatility in reduction conditions allows manufacturers to select the most cost-effective and safe protocol for their specific facility capabilities, ensuring the robust generation of the 3-propylamine-pyrrole intermediate.
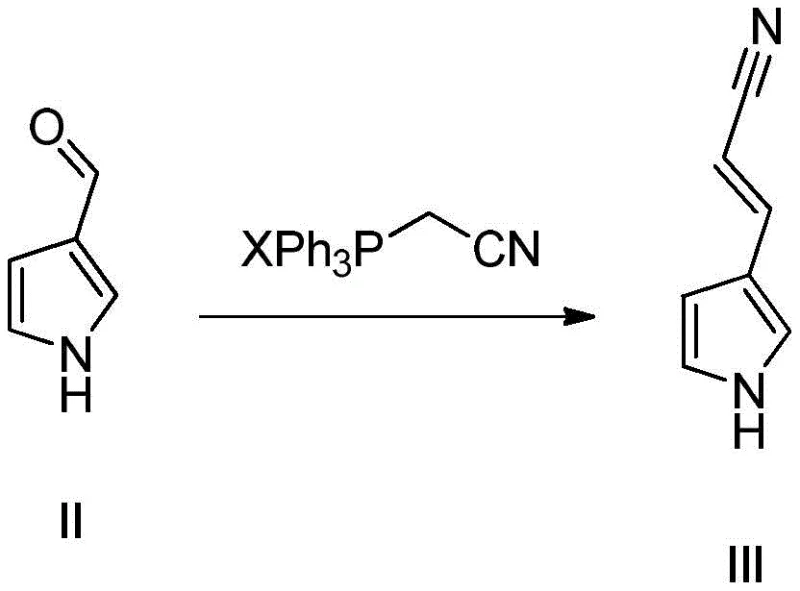
Following the formation of the amine side chain, the synthesis proceeds to the critical coupling stage which defines the structural integrity of the final impurity. To prevent unwanted side reactions during the coupling with the electrophilic fluoroquinolone core, the primary amine of the side chain is first protected using groups such as BOC, tosyl, or trityl. This protection step is vital for chemoselectivity. The subsequent nucleophilic aromatic substitution (SNAr) involves the reaction of the protected amine with 1-cyclopropyl-6,7-difluoro-8-methoxy-1,4-dihydro-4-oxo-3-quinolinecarboxylic acid. In this step, the amine displaces the fluorine atom at the C-7 position of the quinoline ring. The patent highlights the importance of using strong non-nucleophilic bases like potassium tert-butoxide in polar aprotic solvents like DMF to facilitate this displacement efficiently. The reaction temperature is carefully controlled, typically around 80°C, to ensure complete conversion without promoting hydrolysis of the ester or other sensitive functional groups on the quinoline ring. Finally, the protecting group is removed under acidic or hydrogenolytic conditions to reveal the free amine, completing the synthesis of Moxifloxacin degradation impurity J with high structural fidelity.
How to Synthesize Moxifloxacin Degradation Impurity J Efficiently
The synthesis of Moxifloxacin degradation impurity J is a multi-step organic process that requires careful attention to reaction conditions, stoichiometry, and purification techniques to achieve the high purity levels demanded by regulatory agencies. The patented method outlines a clear pathway starting from pyrrole-3-formaldehyde, moving through side-chain elongation and protection, followed by coupling with the quinoline acid derivative, and concluding with deprotection. Each step has been optimized in the patent examples to demonstrate feasibility on a laboratory scale, with specific attention paid to solvent selection, temperature control, and workup procedures to maximize yield. For instance, the Wittig reaction utilizes toluene as a preferred solvent to facilitate the removal of triphenylphosphine oxide byproducts, while the coupling step relies on anhydrous DMF to solubilize the polar quinoline substrate. Understanding these operational nuances is essential for any contract development and manufacturing organization (CDMO) aiming to replicate this process for commercial reference standard production. The detailed standardized synthetic steps for implementing this route are provided in the guide below.
- Perform a Wittig reaction between pyrrole-3-formaldehyde and a triphenyl acetonitrile-based quaternary phosphonium salt under alkaline conditions to form pyrrole-3-acrylonitrile.
- Execute a reduction reaction on the acrylonitrile intermediate to reduce both the C=C double bond and C≡N triple bond, yielding 3-propylamine-pyrrole.
- Protect the amino group of the propylamine-pyrrole using a protecting group such as BOC, tosyl, or trityl to prevent side reactions during coupling.
- Conduct a nucleophilic substitution reaction between the protected amine and 1-cyclopropyl-6,7-difluoro-8-methoxy-1,4-dihydro-4-oxo-3-quinolinecarboxylic acid.
- Remove the amino protecting group via acid hydrolysis or catalytic hydrogenolysis to obtain the final high-purity Moxifloxacin degradation impurity J.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the transition from isolation to total synthesis for Moxifloxacin Impurity J represents a strategic opportunity to optimize costs and secure supply continuity. The traditional isolation method is not only technically challenging but also economically inefficient due to the low recovery rates and the high cost of the starting material (the API itself). In contrast, the synthetic route utilizes commodity chemicals like pyrrole-3-formaldehyde and standard reagents, which are readily available in the global chemical market at stable prices. This shift significantly reduces the raw material cost basis for producing the impurity. Furthermore, the synthetic process is designed with scalability in mind; the reactions involved, such as Wittig olefination and catalytic hydrogenation, are well-understood unit operations that can be easily transferred from laboratory glassware to large-scale industrial reactors without requiring exotic equipment or hazardous high-energy conditions. This ease of scale-up translates directly into improved supply chain reliability, as manufacturers can ramp up production volumes rapidly to meet fluctuating demand from quality control laboratories without the long lead times associated with accumulating sufficient degraded API for isolation.
- Cost Reduction in Manufacturing: The elimination of the need to purchase and degrade expensive Moxifloxacin hydrochloride API results in substantial cost savings. By building the molecule from cheaper precursors, the overall manufacturing cost is drastically lowered. Additionally, the high yields reported in the patent examples mean less waste generation and lower solvent consumption per kilogram of product, further enhancing the economic efficiency of the process. The ability to recycle solvents like toluene and ethyl acetate, which are used in the workup stages, adds another layer of cost optimization that is critical for maintaining competitive pricing in the reference standard market.
- Enhanced Supply Chain Reliability: Relying on a synthetic route decouples the supply of Impurity J from the production schedules of the Moxifloxacin API itself. This independence ensures that reference standard manufacturers can maintain inventory levels regardless of API market fluctuations. The use of robust, high-yielding reactions minimizes the risk of batch failures, which is a common issue with complex isolation procedures. Consequently, customers can expect shorter lead times and more consistent delivery schedules, reducing the risk of stockouts that could delay critical stability testing and regulatory filings for generic drug manufacturers.
- Scalability and Environmental Compliance: The synthetic pathway avoids the use of highly toxic reagents or extreme conditions that would complicate environmental compliance. The waste streams generated, primarily consisting of organic solvents and salt byproducts, are manageable using standard wastewater treatment protocols employed in modern fine chemical facilities. The process is inherently greener than isolation because it avoids the massive solvent usage associated with preparative HPLC purification of trace impurities. This environmental compatibility facilitates easier permitting and operation in regulated jurisdictions, ensuring long-term supply security without the risk of shutdowns due to environmental non-compliance.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of Moxifloxacin degradation impurity J. These answers are derived directly from the technical specifications and experimental data provided in patent CN110627768B, ensuring accuracy and relevance for R&D and quality assurance professionals. Understanding these details is crucial for integrating this reference standard into your analytical workflows and for assessing the feasibility of outsourcing its production to a specialized chemical partner. The information covers aspects of purity, stability, and the specific advantages of the synthetic method over traditional isolation techniques.
Q: Why is synthesizing Moxifloxacin Impurity J superior to isolation?
A: Isolation from photodegraded Moxifloxacin hydrochloride is difficult, yields are low, and purity is hard to control. The patented synthetic method (CN110627768B) allows for the production of Impurity J with purity exceeding 99% and significantly higher yields, ensuring a reliable supply of reference standards for HPLC detection.
Q: What are the critical reaction conditions for the coupling step?
A: The nucleophilic substitution requires anhydrous polar aprotic solvents like DMF or DMSO and strong bases such as potassium tert-butoxide or sodium hydride. The reaction temperature is typically maintained between 80°C and 100°C to ensure complete conversion of the fluoroquinolone core without excessive degradation.
Q: How does this synthesis impact supply chain stability for QC labs?
A: By utilizing readily available starting materials like pyrrole-3-formaldehyde and avoiding the dependency on degrading the final API, manufacturers can produce Impurity J on demand. This eliminates supply bottlenecks associated with isolating trace impurities and ensures consistent batch-to-batch quality for regulatory compliance.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Moxifloxacin Impurity J Supplier
The successful implementation of the synthetic route for Moxifloxacin degradation impurity J requires a partner with deep expertise in complex organic synthesis and a commitment to quality. NINGBO INNO PHARMCHEM stands as a premier CDMO capable of translating such patented methodologies into commercial reality. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that whether you need grams for method validation or kilograms for routine QC, we can deliver. We operate under stringent purity specifications and utilize rigorous QC labs equipped with state-of-the-art analytical instrumentation to verify that every batch of Impurity J meets the >99% purity threshold required for accurate HPLC quantification. Our infrastructure is designed to handle the specific solvent systems and reaction conditions outlined in the patent, guaranteeing a product that is chemically identical to the degradation product found in stability studies.
We invite pharmaceutical companies and analytical laboratories to collaborate with us to secure a stable supply of this critical reference standard. By leveraging our manufacturing capabilities, you can mitigate the risks associated with in-house synthesis or unreliable sourcing. Please contact our technical procurement team to request a Customized Cost-Saving Analysis tailored to your volume requirements. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our optimized process can support your regulatory compliance goals and streamline your quality control operations efficiently.