Scalable Synthesis of Chiral 5-(4-Fluorophenyl)-5-Hydroxypentanoate for Ezetimibe Production
Introduction to Advanced Intermediate Synthesis
The pharmaceutical industry continuously demands more efficient and selective pathways for producing high-value active pharmaceutical ingredients (APIs) and their precursors. Patent CN111138276A introduces a robust synthetic methodology for chiral 5-(4-fluorophenyl)-5-hydroxypentanoate, a critical intermediate in the manufacture of Ezetimibe, a widely prescribed cholesterol absorption inhibitor. This technical disclosure outlines a four-step sequence that begins with readily available commodity chemicals—fluorobenzene and glutaric anhydride—and proceeds through a strategically designed lactone intermediate to achieve the target chiral ester. By circumventing the chemoselectivity issues plaguing conventional direct reduction methods, this process offers a compelling solution for manufacturers seeking to optimize yield and purity profiles. The route demonstrates exceptional control over reaction conditions, utilizing mild temperatures and standard reagents to ensure reproducibility and safety in a production environment.
The strategic value of this synthesis lies in its ability to decouple the reduction and esterification steps, thereby eliminating the risk of over-reduction that frequently compromises batch quality in legacy processes. For R&D directors and process chemists, this represents a significant opportunity to refine impurity profiles and enhance overall process mass intensity (PMI). The method achieves a cumulative yield of up to 83% with purity exceeding 98%, metrics that are highly attractive for commercial supply chains. Furthermore, the reliance on fundamental organic transformations such as Friedel-Crafts acylation and nucleophilic ring-opening ensures that the technology is accessible to a wide range of chemical manufacturing facilities without requiring specialized catalytic infrastructure.
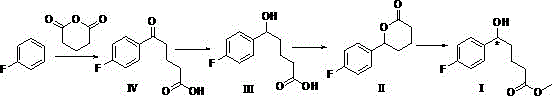
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the preparation of 5-(4-fluorophenyl)-5-hydroxypentanoate derivatives has relied heavily on the direct reduction of 5-(4-fluorophenyl)-5-carbonyl methyl valerate. While conceptually straightforward, this approach suffers from a fundamental lack of chemoselectivity. When strong reducing agents are applied to a molecule containing both a ketone and an ester functionality, there is a persistent thermodynamic drive for the reduction of both groups, leading to the formation of diol byproducts. Controlling this reaction to stop exclusively at the hydroxy-ester stage requires precise and often difficult-to-maintain reaction parameters, such as extremely low temperatures or stoichiometric precision that is hard to replicate on a multi-kilogram scale. Consequently, manufacturers often face challenges with batch-to-batch consistency, lower yields due to over-reduction, and complex purification workflows required to remove closely related alcohol impurities.
The Novel Approach
The methodology described in CN111138276A ingeniously bypasses these selectivity hurdles by altering the oxidation state of the carbon chain during the reduction phase. Instead of attempting to reduce a keto-ester, the process first synthesizes a keto-acid (Compound IV) via Friedel-Crafts acylation. Carboxylic acids are generally inert to sodium borohydride reduction under mild conditions, allowing for the exclusive and high-yielding conversion of the ketone to a secondary alcohol (Compound III) without affecting the acid moiety. Following this selective reduction, the molecule is cyclized into a stable lactone (Compound II), which serves as a protected form of the ester. The final ester functionality is only introduced in the last step via ring-opening with methyllithium. This sequence ensures that the sensitive ester group is never present during the harsh reduction conditions, effectively guaranteeing high purity and simplifying downstream processing.
Mechanistic Insights into Selective Reduction and Lactonization
The core mechanistic advantage of this route is the exploitation of the differential reactivity between carboxylic acids and ketones towards hydride reducing agents. In the second step, sodium borohydride (NaBH4) acts as a nucleophile, delivering a hydride ion to the electrophilic carbonyl carbon of the ketone in Compound IV. Under the specified conditions of -20°C in methanol, the carboxylic acid group remains protonated or forms a stable borate complex that resists reduction, unlike the more electrophilic ketone. This intrinsic selectivity eliminates the need for protecting groups, which would add extra synthetic steps and waste. The subsequent cyclization step utilizes thionyl chloride (SOCl2) to activate the carboxylic acid, forming an acyl chloride intermediate in situ which is immediately attacked by the neighboring hydroxyl group. This intramolecular nucleophilic attack closes the six-membered ring to form the tetrahydropyran-2-one derivative (Compound II), locking the stereochemistry and preparing the molecule for the final functionalization.
Impurity control is further enhanced by the crystallization properties of the intermediates. The patent details specific recrystallization protocols for Compound IV using dichloromethane and for Compound II using ethanol. These purification steps are critical for removing unreacted starting materials and minor side products before they can propagate through the synthesis. For instance, the recrystallization of the lactone intermediate ensures that any residual hydroxy-acid or chlorinated byproducts are removed prior to the sensitive methyllithium reaction. This 'purge point' strategy is essential for maintaining the high purity (>98%) required for pharmaceutical intermediates, ensuring that the final API meets stringent regulatory specifications regarding genotoxic impurities and heavy metals.
How to Synthesize Chiral 5-(4-Fluorophenyl)-5-Hydroxypentanoate Efficiently
Executing this synthesis requires careful attention to thermal management and reagent addition rates, particularly during the exothermic acylation and organolithium steps. The process begins with the generation of the electrophilic acylium ion from glutaric anhydride and aluminum trichloride, followed by the addition of fluorobenzene. Operators must maintain the temperature between -20°C and 0°C to prevent polyacylation or decomposition of the Lewis acid complex. Following the isolation of the keto-acid, the reduction step is performed in methanol, where the controlled addition of sodium borohydride prevents rapid hydrogen evolution and thermal runaway. The detailed standardized synthetic steps for each transformation, including precise molar ratios and workup procedures, are provided in the technical guide below.
- Perform Friedel-Crafts acylation of fluorobenzene with glutaric anhydride using aluminum trichloride to form 5-(4-fluorophenyl)-5-carbonyl valeric acid.
- Reduce the ketone group selectively using sodium borohydride in methanol to yield 5-(4-fluorophenyl)-5-hydroxyvaleric acid.
- Cyclize the hydroxy acid using thionyl chloride to form the lactone intermediate 6-(4-fluorophenyl)-tetrahydropyran-2-one.
- Execute alcoholysis via methyllithium addition at cryogenic temperatures to open the lactone ring and generate the final chiral methyl ester.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, this synthetic route offers substantial advantages rooted in raw material availability and process simplicity. The starting materials, fluorobenzene and glutaric anhydride, are bulk commodity chemicals produced on a massive global scale, ensuring a stable and cost-effective supply base that is not subject to the volatility often seen with specialized chiral building blocks. By avoiding the use of precious metal catalysts (such as palladium or rhodium) or exotic chiral ligands, the process significantly reduces the direct material cost per kilogram. Furthermore, the absence of transition metals simplifies the purification process, as there is no need for expensive metal scavenging resins or rigorous testing for residual heavy metals, which are costly and time-consuming steps in API manufacturing.
- Cost Reduction in Manufacturing: The elimination of protecting group chemistry and the use of inexpensive reducing agents like sodium borohydride contribute to a leaner cost structure. Traditional routes that require ester protection and deprotection add at least two unit operations, increasing solvent consumption, labor hours, and waste disposal costs. By streamlining the synthesis to four direct steps with high yields, the overall operational expenditure is drastically lowered. Additionally, the high selectivity of the reduction step minimizes the loss of valuable intermediates to over-reduced byproducts, maximizing the throughput of the reactor train and improving the overall asset utilization rate for the manufacturing facility.
- Enhanced Supply Chain Reliability: The robustness of the reaction conditions enhances supply continuity. The process operates within a moderate temperature range for most steps (-20°C to 40°C), which can be easily maintained using standard jacketed reactors and glycol cooling systems available in most multipurpose chemical plants. This reduces the dependency on specialized cryogenic infrastructure that might be a bottleneck in some supply chains. Moreover, the intermediates (Compounds IV, III, and II) are stable solids that can be isolated, stored, and quality-controlled before proceeding to the next step. This modularity allows manufacturers to build inventory buffers at intermediate stages, mitigating the risk of total batch loss and ensuring consistent delivery schedules to downstream API producers.
- Scalability and Environmental Compliance: The environmental profile of this synthesis is favorable for large-scale production. The solvents used—dichloromethane, methanol, ethanol, and diethyl ether—are common, recyclable organic solvents with well-established recovery protocols. The avoidance of stoichiometric heavy metal oxidants or reducers reduces the load on wastewater treatment facilities. The high atom economy of the Friedel-Crafts and cyclization steps means less waste generation per unit of product. For supply chain heads focused on sustainability goals and regulatory compliance, this route presents a lower environmental risk footprint compared to older methodologies, facilitating easier permitting and long-term operational viability in regions with strict environmental regulations.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and process descriptions found in the patent literature, providing a clear understanding of the operational requirements and benefits. Understanding these details is crucial for technical teams evaluating the feasibility of technology transfer and for procurement teams assessing the long-term viability of the supply source.
Q: How does this synthesis route improve upon traditional keto-ester reduction methods?
A: Traditional methods often struggle with chemoselectivity, where reducing agents attack both the ketone and the ester group simultaneously. This novel route utilizes a carboxylic acid intermediate which is inherently resistant to borohydride reduction, ensuring exclusive reduction of the ketone moiety before converting to the ester in the final step.
Q: What are the critical temperature controls required for the methyllithium step?
A: The ring-opening alcoholysis reaction requires strict cryogenic conditions, specifically between -60°C and -78°C. Maintaining this low temperature range is essential to control the exothermic nature of the organolithium reaction and prevent side reactions that could compromise the chiral integrity and yield of the final product.
Q: Is this process suitable for large-scale industrial manufacturing?
A: Yes, the process is designed for scalability. It utilizes commodity raw materials like fluorobenzene and glutaric anhydride, avoids expensive transition metal catalysts, and employs standard unit operations such as recrystallization and extraction, making it highly viable for commercial production ranging from hundreds of kilograms to multi-ton scales.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Chiral 5-(4-Fluorophenyl)-5-Hydroxypentanoate Supplier
NINGBO INNO PHARMCHEM stands at the forefront of custom synthesis and contract development, possessing the technical expertise to translate complex patent methodologies into commercial reality. Our team has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to pilot plant and finally to full-scale manufacturing is seamless and efficient. We understand that the production of chiral intermediates like 5-(4-fluorophenyl)-5-hydroxypentanoate requires stringent purity specifications and rigorous QC labs to monitor enantiomeric excess and chemical purity at every stage. Our state-of-the-art facilities are equipped to handle the cryogenic conditions and moisture-sensitive reagents required for the final methyllithium step, guaranteeing a product that meets the highest international pharmacopeial standards.
We invite potential partners to engage with our technical procurement team to discuss how this optimized synthesis route can benefit your specific supply chain. By leveraging our process optimization capabilities, we can provide a Customized Cost-Saving Analysis tailored to your volume requirements, identifying further opportunities for efficiency gains. We encourage you to contact us to request specific COA data from our recent batches and to receive comprehensive route feasibility assessments. Let us collaborate to secure a stable, high-quality supply of this critical Ezetimibe intermediate, driving value and reliability into your pharmaceutical manufacturing operations.