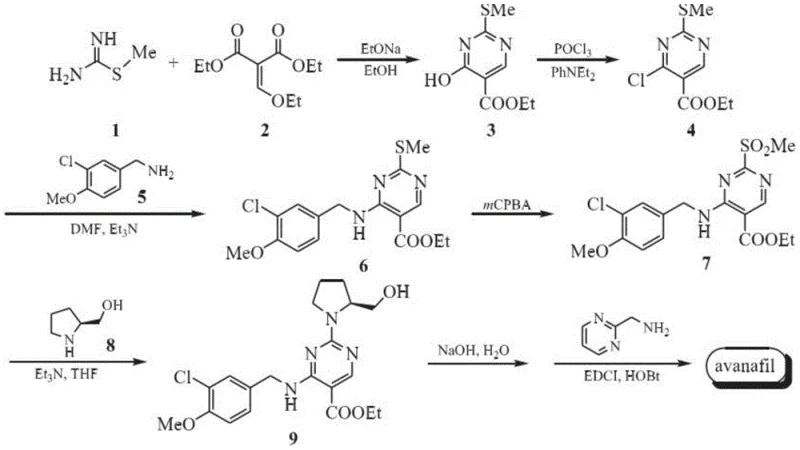
Scalable Preparation of Avanafil Impurity VI for Enhanced Drug Safety and QC
The pharmaceutical industry's relentless pursuit of patient safety has elevated the importance of impurity profiling to a critical regulatory mandate. Patent CN112812101A, published in May 2021, introduces a sophisticated and highly efficient preparation method for a specific Avanafil impurity, designated as Impurity VI. This technical breakthrough addresses a significant gap in the quality control landscape of PDE5 inhibitors, specifically targeting a byproduct that arises during the final condensation steps of Avanafil synthesis. As a leading entity in the fine chemical sector, we recognize that the ability to synthesize reference standards for difficult-to-remove impurities is not merely an academic exercise but a cornerstone of robust supply chain management and regulatory compliance. This patent details a four-step synthetic route that transforms a methylthio-pyrimidine precursor into the target ethylamide impurity with exceptional purity, providing pharmaceutical manufacturers with the essential tools to validate their analytical methods and ensure the safety of the final drug product.
The strategic value of this technology lies in its direct application to the commercial scale-up of complex pharmaceutical intermediates. By establishing a reliable source for Impurity VI, manufacturers can better understand the degradation pathways of Avanafil and implement more effective purification strategies. This report delves into the mechanistic nuances of this novel pathway, contrasting it with conventional limitations, and outlines the tangible commercial benefits for procurement and supply chain stakeholders seeking to optimize their API production workflows.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
In the traditional manufacturing landscape of Avanafil, the formation of Impurity VI is often an unintended and problematic consequence of the final amidation step. As disclosed in prior art such as US6797709, the synthesis typically employs condensing agents like EDCI (1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride). Under certain reaction conditions, particularly in the presence of moisture, EDCI can decompose to release ethylamine. This free ethylamine acts as a nucleophile, attacking the activated ester intermediate (Compound IV in the context of the main API synthesis) to form the ethylamide derivative, which is structurally identical to Impurity VI. The critical issue here is that this impurity is generated in situ in trace amounts, making its isolation for characterization extremely difficult and its quantification in the final API challenging without an authentic reference standard. The lack of a dedicated synthesis route for this specific impurity has historically hindered the ability of quality control laboratories to set precise acceptance criteria, potentially compromising the safety profile of the generic drug versions entering the market.
The Novel Approach
The methodology presented in CN112812101A fundamentally shifts the paradigm from accidental formation to intentional, controlled synthesis. Instead of relying on the stochastic decomposition of coupling reagents, this novel approach constructs Impurity VI through a deliberate, linear sequence starting from a stable pyrimidine scaffold. The process begins with the selective oxidation of a sulfide to a sulfoxide, followed by a stereoselective substitution with L-prolinol, hydrolysis of the ester moiety, and finally, a targeted condensation with ethylamine hydrochloride. This route ensures that the impurity is produced as a discrete, high-purity chemical entity rather than a trace contaminant. By decoupling the impurity synthesis from the main API production line, manufacturers gain the ability to produce gram-to-kilogram quantities of the reference standard independently. This independence is crucial for reducing lead time for high-purity pharmaceutical intermediates required for method validation, allowing R&D teams to proceed with stability studies and regulatory filings without waiting for impurity isolation from failed API batches.
Mechanistic Insights into the Oxidation and Condensation Strategy
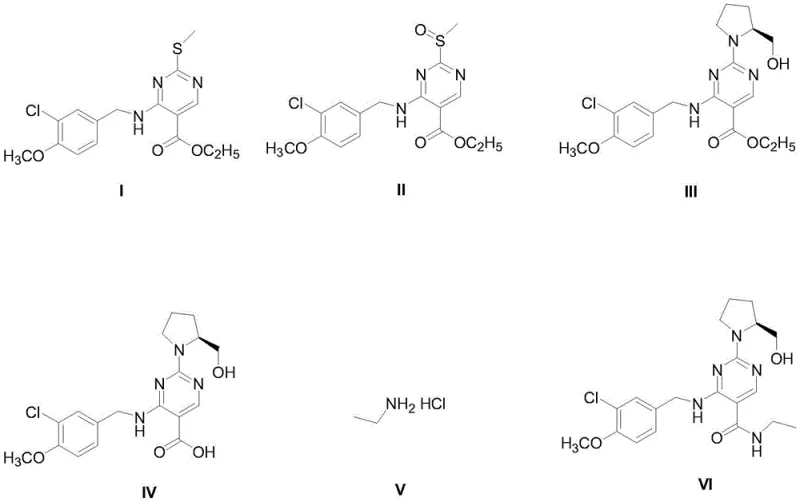
The core of this synthetic innovation lies in the precise control of oxidation states and nucleophilic substitutions. The first step involves the transformation of 4-(3-chloro-4-methoxy phenylamino)-5-ethoxycarbonyl-2-methylthiopyrimidine (Compound I) into its corresponding sulfoxide (Compound II). The patent specifies the use of oxidants such as 30% hydrogen peroxide or m-chloroperoxybenzoic acid (mCPBA) in solvents like toluene or dichloromethane. The reaction is conducted at mild temperatures ranging from 25°C to 50°C, with a preferred embodiment at 40°C. This thermal window is critical; it is high enough to drive the oxidation to completion within 24 hours but low enough to prevent over-oxidation to the sulfone or degradation of the sensitive pyrimidine ring. The stoichiometry is carefully balanced, with a molar ratio of substrate to oxidant preferably set at 1:4, ensuring complete conversion while minimizing excess oxidant that could complicate downstream purification. This controlled oxidation sets the stage for the subsequent nucleophilic attack by activating the C2 position of the pyrimidine ring.
Following oxidation, the process moves to a nucleophilic aromatic substitution where the methylsulfinyl group is displaced by L-prolinol. This step is facilitated by organic bases such as pyridine or triethylamine in dichloromethane. The stereochemistry of the L-prolinol is preserved, which is vital as the final impurity must match the stereochemical profile of the impurity formed in the chiral API synthesis. The subsequent hydrolysis of the ethyl ester to the carboxylic acid (Compound IV) is achieved using alkali metal carbonates like potassium carbonate in methanol. This saponification is highly efficient, with yields reported up to 96.9%, demonstrating the robustness of the ester cleavage under these mild basic conditions. Finally, the carboxylic acid is activated using EDCI and coupled with ethylamine hydrochloride. Unlike the uncontrolled formation in the API process, here the ethylamine is introduced as a stoichiometric reagent (molar ratio 1:1.5), driving the reaction to high conversion (yields up to 96.4%) and resulting in a final product with HPLC purity exceeding 98.75%. This level of purity is essential for its role as a quantitative reference standard.
How to Synthesize Avanafil Impurity VI Efficiently
The synthesis of Avanafil Impurity VI described in this patent offers a reproducible and scalable protocol suitable for both laboratory reference standard production and larger-scale intermediate manufacturing. The process is designed to minimize operational complexity while maximizing yield and purity, making it an ideal candidate for technology transfer. The following guide outlines the critical operational parameters derived from the patent examples, focusing on the optimal conditions identified for each transformation step. For detailed, step-by-step instructions including specific reagent grades and workup procedures, please refer to the standardized protocol below.
- Oxidize 4-(3-chloro-4-methoxy phenylamino)-5-ethoxycarbonyl-2-methylthiopyrimidine (Compound I) using an oxidant like 30% hydrogen peroxide in toluene at 40°C to obtain Compound II.
- React Compound II with L-prolinol in dichloromethane using pyridine as a base to perform nucleophilic substitution, yielding Compound III.
- Hydrolyze the ester group of Compound III using potassium carbonate in methanol to generate the carboxylic acid intermediate, Compound IV.
- Condense Compound IV with ethylamine hydrochloride (Compound V) using EDCI as a coupling agent in DMA solvent to finalize the synthesis of Avanafil Impurity VI.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented synthesis route offers distinct strategic advantages beyond mere technical feasibility. The primary value proposition is the stabilization of the supply chain for critical quality control materials. By utilizing a dedicated synthesis route for Impurity VI, companies eliminate the dependency on isolating trace impurities from expensive API batches, which is a costly and unpredictable process. This shift allows for the independent sourcing or manufacturing of the impurity standard, thereby significantly reducing the risk of supply disruption during critical phases of drug development and regulatory audit. Furthermore, the use of commodity chemicals such as hydrogen peroxide, L-prolinol, and ethylamine hydrochloride ensures that raw material costs remain stable and predictable, shielding the project from the volatility associated with specialized reagents.
- Cost Reduction in Manufacturing: The process described eliminates the need for complex chromatographic separations often required to isolate trace impurities from reaction mixtures. Since the synthesis is designed to produce the impurity as the major product with high selectivity, purification is simplified to standard crystallization and washing steps. This reduction in downstream processing complexity translates directly into lower manufacturing costs. Additionally, the avoidance of exotic catalysts and the use of recyclable solvents like toluene and dichloromethane further enhance the economic viability of the process, offering substantial cost savings in the production of high-value reference standards.
- Enhanced Supply Chain Reliability: The synthetic route relies on widely available starting materials and reagents that are not subject to strict geopolitical trade restrictions or single-source bottlenecks. The robustness of the reaction conditions—operating at ambient to mild temperatures—means that the process can be executed in a wide range of manufacturing facilities without requiring specialized cryogenic or high-pressure equipment. This flexibility enhances supply chain resilience, allowing for multi-site production capabilities and ensuring continuous availability of the impurity standard to support global quality control operations.
- Scalability and Environmental Compliance: From an environmental and safety perspective, the process is favorable for scale-up. The oxidation step uses hydrogen peroxide, which generates water as a byproduct, aligning with green chemistry principles. The absence of heavy metal catalysts removes the need for expensive and waste-generating metal scavenging steps. The overall atom economy is improved by the high yields reported in the patent examples (often exceeding 90% in key steps), which minimizes waste generation. These factors collectively simplify the environmental permitting process for large-scale production and reduce the overall environmental footprint of the manufacturing operation.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis and application of Avanafil Impurity VI. These answers are derived directly from the experimental data and technical specifications provided in patent CN112812101A, ensuring accuracy and relevance for R&D and quality assurance professionals. Understanding these details is crucial for integrating this impurity standard into your existing analytical frameworks and supply chain strategies.
Q: Why is the synthesis of Avanafil Impurity VI critical for drug safety?
A: During the commercial synthesis of Avanafil using EDCI condensation agents, trace amounts of ethylamine can be generated via decomposition. This ethylamine reacts with intermediates to form Impurity VI, which is difficult to remove. Having a reference standard of Impurity VI allows manufacturers to accurately quantify and control this specific genotoxic potential, ensuring the final API meets stringent regulatory safety profiles.
Q: What are the key advantages of the oxidation method described in CN112812101A?
A: The patent describes a highly efficient oxidation step converting the methylthio group to a sulfoxide (Compound I to II) using 30% hydrogen peroxide. This method operates at mild temperatures (25-50°C) and achieves high yields (up to 81.2%), avoiding the use of hazardous heavy metal oxidants and simplifying the workup procedure compared to traditional methods.
Q: How does this process support commercial scale-up for pharmaceutical intermediates?
A: The process utilizes common, scalable solvents such as toluene, dichloromethane, and methanol, and avoids cryogenic conditions. The reactions proceed at room temperature or mild heating (40°C), making the thermal load manageable for large-scale reactors. Furthermore, the purification steps involve standard aqueous washes and crystallization, which are easily transferable from pilot plant to multi-ton production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Avanafil Impurity VI Supplier
At NINGBO INNO PHARMCHEM, we understand that the integrity of your pharmaceutical products depends on the quality of every component, including the impurities used for validation. Our team of expert chemists has extensively analyzed the technology disclosed in CN112812101A and is fully prepared to leverage this knowledge for your benefit. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that whether you need milligrams for analytical method development or kilograms for stability testing, we can deliver with consistency. Our state-of-the-art rigorous QC labs and commitment to stringent purity specifications guarantee that every batch of Avanafil Impurity VI we supply meets the highest international standards, providing you with the confidence needed for successful regulatory submissions.
We invite you to collaborate with us to optimize your supply chain for Avanafil-related materials. By partnering with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements and timeline. We encourage you to reach out today to discuss your project needs,索取 specific COA data for our available batches, and receive comprehensive route feasibility assessments. Let us be your strategic partner in navigating the complexities of pharmaceutical intermediate manufacturing, ensuring your path to market is smooth, compliant, and cost-effective.