Mastering Gabapentin Impurity Control: Novel Synthesis of Compound VII for Global QC Standards
Mastering Gabapentin Impurity Control: Novel Synthesis of Compound VII for Global QC Standards
In the highly regulated landscape of antiepileptic drug manufacturing, the identification and control of trace impurities are paramount for patient safety and regulatory compliance. Patent CN113698339A, published in late 2021, introduces a significant breakthrough in the quality assurance of Gabapentin by detailing the isolation and synthesis of a previously unknown degradation product, designated as Impurity VII (2-[1-[(1,3-dioxa-2-azaspiro[4.5]decane-2-yl)methyl]cyclohexyl]-acetic acid). This specific compound, characterized by a complex spiro-succinimide structure, was identified as a critical stability indicator that naturally accumulates in finished Gabapentin products over time, posing potential risks if left unmonitored. The patent provides a robust, scalable synthetic route to produce this impurity as a high-purity reference standard, enabling pharmaceutical manufacturers to implement precise quantitative analysis methods such as HPLC with external calibration. By mastering the synthesis of this specific marker, producers can transition from reactive quality control to proactive stability management, ensuring that their bulk drug substances meet the stringent purity specifications required by global health authorities.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditionally, the monitoring of Gabapentin impurities has relied heavily on area normalization methods or non-specific detection techniques that often fail to distinguish between structurally similar degradation products. In standard manufacturing workflows, Impurity VII arises unpredictably during the long-term storage of the active pharmaceutical ingredient (API), particularly when trace amounts of its dicarboxylic acid precursor remain in the matrix. Without an authentic reference standard, quality control laboratories are forced to estimate the content of this impurity based on relative retention times (RRT) and assumed response factors, which frequently leads to significant inaccuracies. For instance, the patent data reveals that using area normalization can underestimate the main component's purity because Impurity VII exhibits a stronger detector response than Gabapentin itself. This analytical blind spot creates a dangerous scenario where a batch might pass release criteria despite containing elevated levels of a genotoxic or unstable degradant, ultimately leading to potential recalls or regulatory citations during audits. Furthermore, the inability to synthetically reproduce this impurity hinders root cause analysis, leaving process chemists unable to pinpoint exactly which upstream reaction conditions are promoting its formation.
The Novel Approach
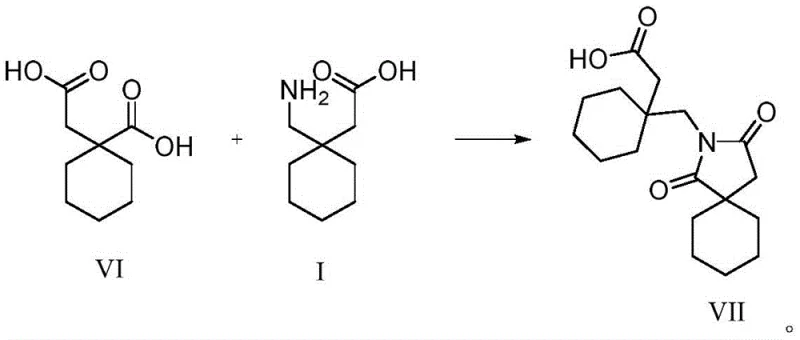
The methodology disclosed in CN113698339A revolutionizes this quality control paradigm by offering a direct, high-yield synthesis of Impurity VII, transforming it from an unknown variable into a quantifiable standard. The core innovation lies in the biomimetic condensation of Gabapentin (I) with (1-carboxymethyl)cyclohexylcarboxylic acid (VI) under controlled thermal conditions. Unlike harsh degradation studies that produce complex mixtures of unknown byproducts, this targeted approach utilizes a specific stoichiometric reaction in aqueous or alcoholic media to drive the formation of the spiro-imide ring with exceptional selectivity. By intentionally synthesizing the impurity, manufacturers gain the ability to spike validation samples with known concentrations, thereby calibrating their HPLC methods to detect even trace amounts (e.g., below 0.1%) with high precision. This shift allows for the implementation of the external standard method, which corrects for response factor discrepancies and provides a true reflection of product purity. Consequently, this approach not only satisfies rigorous ICH Q3 guidelines for impurity qualification but also empowers supply chain teams to set scientifically justified specification limits that protect product integrity throughout its shelf life.
Mechanistic Insights into Spiro-Imide Cyclization
The formation of Impurity VII represents a fascinating example of thermal dehydration and cyclization chemistry occurring within the solid state or solution phase of the API. Mechanistically, the reaction initiates when the primary amine group of Gabapentin nucleophilically attacks one of the carboxylic acid moieties of the precursor (VI), forming an initial amide bond. Under the influence of heat (30-100°C) and the removal of water, the second carboxylic acid group undergoes an intramolecular attack on the amide nitrogen or adjacent carbonyl, closing the five-membered succinimide ring. This cyclization creates the sterically hindered spiro[4.5]decane system, which is thermodynamically stable and resistant to further hydrolysis under normal storage conditions. The patent highlights that this transformation is facilitated by the spatial proximity of the functional groups in the transition state, suggesting that even in the solid state, molecular mobility allows for this slow degradation over months of storage. Understanding this mechanism is crucial for R&D directors, as it indicates that minimizing the residual levels of the dicarboxylic acid precursor (VI) in the final Gabapentin crystallization step is the most effective strategy to prevent Impurity VII generation at the source.
Furthermore, the structural elucidation provided by LC-HRMS and NMR spectroscopy confirms the unique electronic environment of the spiro-center, which influences the compound's chromatographic behavior. The presence of two carbonyl groups in the imide ring creates a polar region that interacts distinctly with reverse-phase C18 columns, resulting in a Relative Retention Time (RRT) of approximately 0.84 relative to Gabapentin. This specific elution profile is critical for method development, as it ensures the impurity is well-resolved from the main peak and other common process-related impurities. The detailed NMR data, including characteristic proton shifts for the methylene protons adjacent to the nitrogen and the spiro-carbon, serves as a fingerprint for identity confirmation. For analytical chemists, this level of structural detail validates the specificity of the new QC method, ensuring that the peak observed at RRT 0.84 is indeed the stability-indicating impurity and not an artifact or a different isomer, thereby eliminating false positives in stability reporting.
How to Synthesize Gabapentin Impurity VII Efficiently
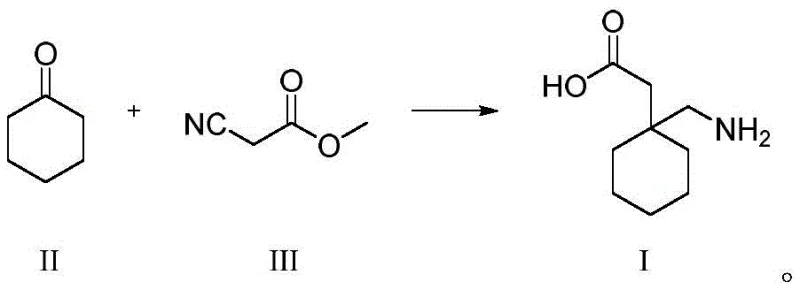
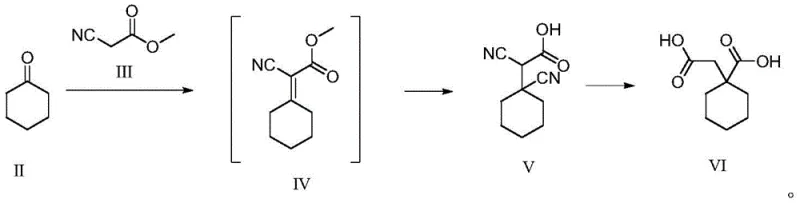
The synthesis of this critical reference standard is designed for operational simplicity and scalability, utilizing readily available starting materials and benign solvent systems. The process begins with the preparation of the key intermediate, (1-carboxymethyl)cyclohexylcarboxylic acid (VI), which is synthesized from cyclohexanone and methyl cyanoacetate through a Knoevenagel condensation followed by cyanation and hydrolysis. Once the precursor is secured, the final step involves a straightforward condensation with Gabapentin in water or lower alcohols. The reaction conditions are mild, requiring temperatures between 30°C and 100°C, making it accessible for standard pilot plant equipment without the need for specialized high-pressure reactors. This accessibility ensures that reference material production can be easily scaled from gram quantities for lab validation to kilogram batches for multi-site QC distribution. For detailed operational parameters, stoichiometry, and workup procedures, please refer to the standardized synthesis guide below.
- Preparation of Precursor VI: React cyclohexanone with methyl cyanoacetate followed by cyanation and hydrolysis to obtain (1-carboxymethyl)cyclohexylcarboxylic acid.
- Condensation Reaction: Mix Precursor VI with Gabapentin (I) in an aqueous or alcoholic solvent system.
- Thermal Cyclization: Heat the mixture to 30-100°C for 3-10 hours to facilitate dehydration and spiro-imide ring formation, yielding Impurity VII.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain leaders, the ability to source or internally produce Impurity VII offers substantial strategic advantages beyond mere regulatory compliance. The primary value proposition lies in the drastic reduction of risk associated with product recalls and market withdrawals. By possessing a reliable supply of this reference standard, companies can validate their stability data with absolute certainty, preventing the release of batches that might fail prematurely in the market due to undetected impurity growth. This proactive quality assurance translates directly into cost avoidance, as the financial impact of a recall far exceeds the investment in developing robust analytical controls. Moreover, the synthesis route described avoids the use of exotic catalysts or hazardous reagents, relying instead on commodity chemicals like cyclohexanone and simple acids. This reliance on common feedstocks ensures that the production of the reference standard is not subject to the volatile supply chains often associated with specialized fine chemicals, guaranteeing continuity of supply for QC laboratories globally.
- Cost Reduction in Manufacturing: The synthesis protocol eliminates the need for expensive chromatographic purification steps typically required to isolate trace impurities from degradation mixtures. By driving the reaction to high conversion using stoichiometric excesses of inexpensive starting materials, the process yields the target impurity as a crystalline solid that can be purified via simple recrystallization. This streamlined downstream processing significantly lowers the cost per gram of the reference standard compared to purchasing certified materials from third-party vendors, who often charge premium prices for low-volume specialty compounds. Additionally, the high atom economy of the condensation reaction minimizes waste generation, reducing disposal costs and aligning with green chemistry initiatives that are increasingly important for corporate sustainability goals.
- Enhanced Supply Chain Reliability: Dependence on external suppliers for critical reference standards creates a single point of failure in the quality control workflow; if the vendor faces production issues, the entire batch release process can be halted. By mastering the in-house or local contract synthesis of Impurity VII, pharmaceutical companies insulate themselves from these external disruptions. The raw materials for this synthesis, such as Gabapentin and cyclohexanone derivatives, are produced on a multi-ton scale globally, ensuring that feedstock availability is never a bottleneck. This self-sufficiency allows QA departments to maintain uninterrupted testing schedules, ensuring that finished products reach patients without delay, thereby strengthening the overall resilience of the pharmaceutical supply network against global logistical shocks.
- Scalability and Environmental Compliance: The reaction conditions described in the patent are inherently scalable, utilizing water or ethanol as primary solvents, which are environmentally benign and easy to recover. Unlike processes requiring chlorinated solvents or heavy metal catalysts, this aqueous-based condensation generates wastewater that is easier to treat and dispose of in compliance with environmental regulations. The absence of toxic reagents simplifies the permitting process for manufacturing facilities and reduces the burden on EHS (Environment, Health, and Safety) teams. Furthermore, the robustness of the reaction allows for easy scale-up from laboratory glassware to industrial reactors without significant re-optimization, facilitating the rapid production of large quantities of reference material needed for multinational clinical trials or routine commercial batch testing.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis and application of Gabapentin Impurity VII. These insights are derived directly from the experimental data and stability studies presented in the patent literature, providing a factual basis for decision-making. Understanding the nuances of this impurity's behavior is essential for both analytical scientists developing methods and executives managing product lifecycles. We encourage stakeholders to review these details to fully appreciate the value of integrating this reference standard into their quality systems.
Q: Why is Impurity VII critical for Gabapentin stability testing?
A: Impurity VII is a degradation product that forms during storage, increasing over time and compromising product safety. Having a pure reference standard allows manufacturers to accurately quantify this specific risk and validate shelf-life stability according to ICH guidelines.
Q: What is the chemical mechanism behind the formation of Impurity VII?
A: It forms via a double condensation reaction between the primary amine of Gabapentin and the dicarboxylic acid precursor (VI), resulting in a stable spiro-succinimide ring structure through dehydration.
Q: Can this impurity be removed from the final API?
A: While difficult to remove once formed due to structural similarity, controlling the levels of precursor VI in the starting material and optimizing storage conditions can significantly inhibit its generation during the product lifecycle.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Gabapentin Impurity VII Supplier
At NINGBO INNO PHARMCHEM, we understand that the integrity of your pharmaceutical products relies on the precision of your analytical controls. As a leading CDMO and fine chemical manufacturer, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that even complex molecules like spiro-imide impurities are synthesized with the highest fidelity. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every milligram of reference standard we provide is fully characterized by NMR, MS, and HPLC. We are committed to supporting your regulatory filings with documentation that stands up to the scrutiny of global health authorities, helping you maintain the highest standards of patient safety.
We invite you to collaborate with our technical team to optimize your impurity control strategies. Whether you require custom synthesis of hard-to-find degradation products or a Customized Cost-Saving Analysis for your existing supply chain, we are ready to assist. Please contact our technical procurement team today to request specific COA data, route feasibility assessments, and competitive quotations that will empower your quality assurance operations.