Advanced Synthesis of Fluorine-Containing 1,1-Stilbene Derivatives for Oncology Drug Development
Advanced Synthesis of Fluorine-Containing 1,1-Stilbene Derivatives for Oncology Drug Development
The pharmaceutical industry is constantly seeking next-generation antitumor agents that overcome the pharmacokinetic limitations of natural products. Patent CN108276267B introduces a groundbreaking class of fluorine-containing 1,1-stilbene derivatives designed to address the instability issues inherent in traditional microtubule destabilizing agents. The reference compound, Combretastatin A-4 (CA-4), shown in 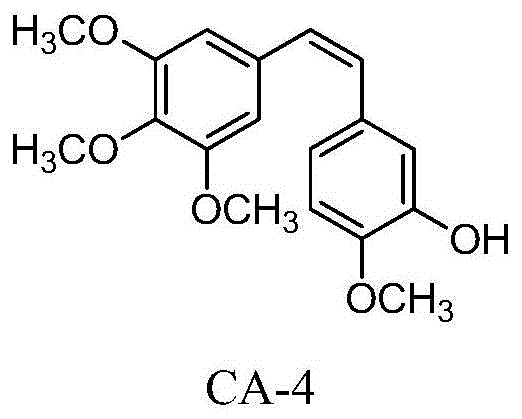 , is a potent vascular disrupting agent but suffers from rapid isomerization from the active cis-form to the inactive trans-form. This patent details a sophisticated chemical strategy where specific positions on the aromatic rings are modified with fluorine atoms and other substituents to lock the bioactive conformation. By acting as a reliable pharmaceutical intermediate supplier, we recognize that such structural innovations are critical for developing drugs with improved metabolic profiles and enhanced therapeutic windows.
, is a potent vascular disrupting agent but suffers from rapid isomerization from the active cis-form to the inactive trans-form. This patent details a sophisticated chemical strategy where specific positions on the aromatic rings are modified with fluorine atoms and other substituents to lock the bioactive conformation. By acting as a reliable pharmaceutical intermediate supplier, we recognize that such structural innovations are critical for developing drugs with improved metabolic profiles and enhanced therapeutic windows.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthesis of stilbene-based antitumor agents often relies on Wittig or Horner-Wadsworth-Emmons reactions which can struggle with stereoselectivity, frequently yielding mixtures of cis and trans isomers that require difficult and costly separation processes. Furthermore, the native stilbene double bond is susceptible to metabolic oxidation and isomerization in physiological conditions, leading to a short half-life and reduced efficacy in vivo. The lack of metabolic stability in early-generation CA-4 analogues has been a significant bottleneck in clinical translation, necessitating complex prodrug strategies or structural rigidification that often complicates the manufacturing process. These conventional pathways often involve harsh conditions or expensive chiral catalysts that drive up the cost of goods, making them less attractive for generic API manufacturing or large-scale commercial production.
The Novel Approach
The novel approach described in the patent utilizes a gem-difluoro methylene group to replace the labile double bond or adjacent carbonyl functionalities, effectively acting as a bioisostere that mimics the transition state while resisting metabolic degradation. As illustrated in the general formula (I) in 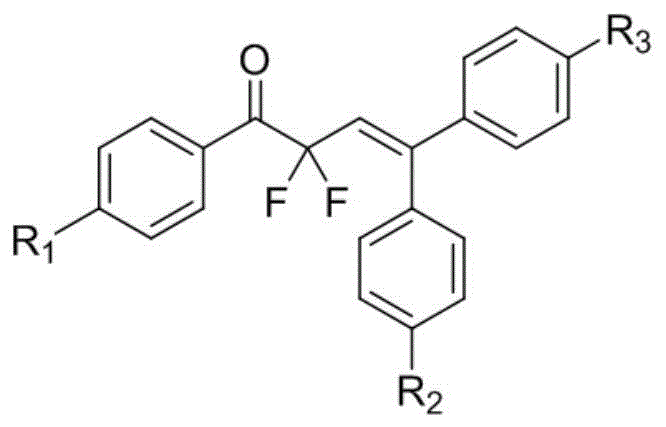 , the strategic placement of fluorine atoms at the alpha-position of the ketone moiety creates a strong electron-withdrawing effect that stabilizes the molecule against enzymatic attack. This method allows for the modular introduction of diverse substituents (R1, R2, R3) such as methyl, cyano, or halogens, enabling fine-tuning of the physicochemical properties like solubility and lipophilicity. This flexibility is paramount for a reliable pharmaceutical intermediate supplier aiming to optimize lead compounds for specific tumor targets without redesigning the entire synthetic route.
, the strategic placement of fluorine atoms at the alpha-position of the ketone moiety creates a strong electron-withdrawing effect that stabilizes the molecule against enzymatic attack. This method allows for the modular introduction of diverse substituents (R1, R2, R3) such as methyl, cyano, or halogens, enabling fine-tuning of the physicochemical properties like solubility and lipophilicity. This flexibility is paramount for a reliable pharmaceutical intermediate supplier aiming to optimize lead compounds for specific tumor targets without redesigning the entire synthetic route.
Mechanistic Insights into Fluorination and Cross-Coupling Strategies
The core of this synthesis lies in the construction of the fluorinated ketone backbone followed by carbon-carbon bond formation. The process begins with a Claisen condensation between a substituted acetophenone and ethyl trifluoroacetate under basic conditions, generating a beta-diketone intermediate. This intermediate is then subjected to selective fluorination using Selectfluor, a powerful electrophilic fluorinating agent that ensures high regioselectivity. Following fluorination, the material undergoes iodination in the presence of lithium bromide and triethylamine to install a leaving group necessary for subsequent coupling. The synthesis of the key intermediate (A), depicted in 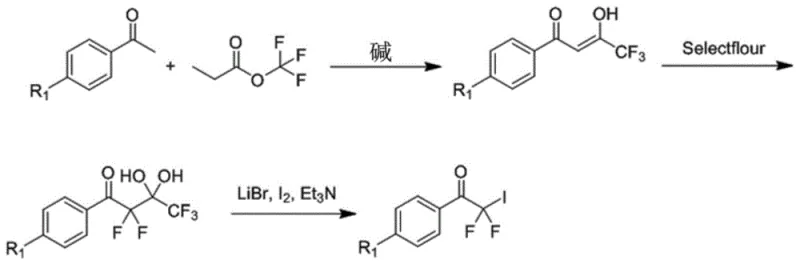 , highlights the precision required to install the gem-difluoro motif without compromising the integrity of the aromatic rings. This step is crucial as it sets the stage for the radical addition reaction that builds the stilbene skeleton.
, highlights the precision required to install the gem-difluoro motif without compromising the integrity of the aromatic rings. This step is crucial as it sets the stage for the radical addition reaction that builds the stilbene skeleton.
The final assembly of the target molecule involves a radical addition reaction between the iodinated ketone and a substituted phenylacetylene, initiated by AIBN under inert atmosphere. This forms the vinyl-iodide intermediate which is then coupled with various phenylboronic acids via a palladium-catalyzed Suzuki-Miyaura cross-coupling reaction. The complete synthetic route to the final fluorine-containing 1,1-stilbene derivative is shown in 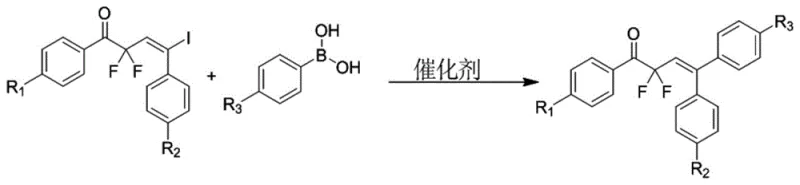 . The use of PdCl2(PPh3)2 as the catalyst ensures efficient coupling even with sterically hindered substrates. From a mechanistic standpoint, the electron-deficient nature of the fluorinated ketone facilitates the radical addition step, while the mild conditions of the Suzuki coupling preserve the sensitive functional groups on the aromatic rings. This robust mechanism ensures high purity and minimizes the formation of difficult-to-remove impurities, a key concern for R&D directors focusing on impurity profiles.
. The use of PdCl2(PPh3)2 as the catalyst ensures efficient coupling even with sterically hindered substrates. From a mechanistic standpoint, the electron-deficient nature of the fluorinated ketone facilitates the radical addition step, while the mild conditions of the Suzuki coupling preserve the sensitive functional groups on the aromatic rings. This robust mechanism ensures high purity and minimizes the formation of difficult-to-remove impurities, a key concern for R&D directors focusing on impurity profiles.
How to Synthesize Fluorine-Containing 1,1-Stilbene Derivatives Efficiently
The synthesis of these high-value intermediates requires precise control over reaction parameters to maximize yield and purity. The patented process outlines a streamlined sequence that avoids unnecessary protection-deprotection steps, thereby reducing waste and processing time. The initial condensation and fluorination steps are performed in standard reactor vessels, while the radical addition requires careful temperature control between 55-65°C to manage the exotherm and ensure safety. The final purification typically involves column chromatography or recrystallization, depending on the specific substituents used. For detailed operational parameters, stoichiometry, and workup procedures, please refer to the standardized guide below.
- Perform Claisen condensation between substituted acetophenone and ethyl trifluoroacetate under basic conditions to form the beta-diketone intermediate.
- Execute selective fluorination using Selectfluor followed by iodination with LiBr and I2 to generate the gem-difluoro iodo-ketone precursor.
- Conduct radical addition with phenylacetylene using AIBN initiator, followed by a final Pd-catalyzed Suzuki coupling with phenylboronic acid to yield the target stilbene derivative.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this fluorinated synthetic route offers distinct logistical and economic advantages over traditional stilbene synthesis. The reliance on commodity chemicals such as acetophenones, phenylacetylenes, and boronic acids ensures a stable and diversified supply base, mitigating the risk of raw material shortages. Furthermore, the elimination of complex chiral resolution steps significantly simplifies the manufacturing workflow, leading to substantial cost savings in labor and solvent consumption. The robustness of the radical addition and Suzuki coupling steps means that the process is highly tolerant to minor variations in reaction conditions, which translates to consistent batch-to-batch quality and reduced rejection rates in a GMP environment.
- Cost Reduction in Manufacturing: The synthetic pathway eliminates the need for expensive chiral catalysts or difficult separations of cis/trans isomers, which are major cost drivers in conventional stilbene production. By utilizing a gem-difluoro strategy, the molecule is inherently stabilized, removing the downstream costs associated with stabilizing formulations or frequent dosing. The use of widely available reagents like Selectfluor and standard palladium catalysts allows for competitive pricing models, ensuring that cost reduction in API manufacturing is achieved without compromising on the quality of the active pharmaceutical ingredient.
- Enhanced Supply Chain Reliability: The modular nature of the synthesis, where different R-groups can be swapped in the final coupling step, allows for the rapid production of a library of analogues from a common advanced intermediate. This flexibility means that inventory risks are minimized, as a single stock of the fluorinated ketone precursor can serve multiple product lines. Additionally, the reaction conditions do not require cryogenic temperatures or ultra-high pressures, making the process compatible with existing multipurpose reactors in most contract manufacturing organizations, thereby reducing lead time for high-purity pharmaceutical intermediates.
- Scalability and Environmental Compliance: The process generates minimal hazardous waste compared to older methods that might utilize heavy metal oxidants or toxic phosphine byproducts. The workup procedures involve standard aqueous extractions and organic solvent washes, which are easily managed by existing wastewater treatment facilities. This environmental compatibility facilitates easier regulatory approval and supports sustainable manufacturing goals. The scalability is further evidenced by the use of radical initiators and thermal coupling reactions which translate linearly from gram-scale R&D to multi-tonne commercial production, ensuring a continuous supply for clinical and market needs.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these fluorinated stilbene derivatives. The answers are derived directly from the experimental data and technical specifications outlined in the patent documentation, ensuring accuracy for our partners in the pharmaceutical sector.
Q: What is the primary advantage of introducing fluorine atoms into the stilbene scaffold?
A: The introduction of fluorine atoms, particularly in a gem-difluoro configuration, significantly enhances metabolic stability and lipophilicity without drastically altering molecular volume. This modification helps lock the conformation, preventing the unfavorable cis-trans isomerization often seen in native Combretastatin A-4, thereby maintaining potent tubulin polymerization inhibition.
Q: Is this synthetic route suitable for large-scale commercial production?
A: Yes, the route utilizes robust and commercially available starting materials such as substituted acetophenones and phenylacetylenes. The reaction conditions, including the use of standard radical initiators like AIBN and palladium catalysts for cross-coupling, are well-established in industrial settings, facilitating easy scale-up from kilogram to tonne levels.
Q: How does the biological activity of these derivatives compare to CA-4?
A: In vitro evaluations demonstrate that specific fluorine-containing derivatives exhibit broad-spectrum antitumor activity comparable to, and in some cases exceeding, the positive control CA-4. For instance, certain analogues showed superior IC50 values against Hela and MGC803 cell lines, indicating that the electronic effects of the fluorine substituents effectively enhance binding affinity at the colchicine site.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Fluorine-Containing 1,1-Stilbene Derivative Supplier
At NINGBO INNO PHARMCHEM, we understand that the transition from a patented laboratory method to a commercial reality requires deep technical expertise and robust infrastructure. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with consistency and reliability. We adhere to stringent purity specifications and operate rigorous QC labs equipped with advanced analytical instrumentation to verify the identity and potency of every batch. Our commitment to quality assurance means that every fluorine-containing intermediate we deliver meets the highest global standards for oncology drug development.
We invite you to collaborate with us to leverage this innovative technology for your pipeline. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and route feasibility assessments to help you accelerate your project timelines. Let us be your partner in bringing these potent antitumor agents from the bench to the bedside.