Advanced Synthesis of Tenofovir Phenyl Ester Intermediates for Commercial API Production
Advanced Synthesis of Tenofovir Phenyl Ester Intermediates for Commercial API Production
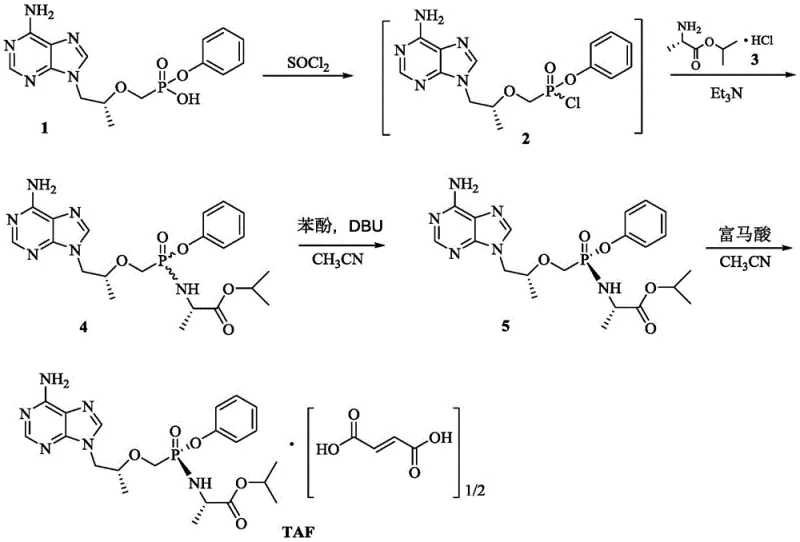
The pharmaceutical industry continuously seeks robust manufacturing pathways for critical antiviral agents, particularly for Tenofovir Alafenamide (TAF), a cornerstone treatment for chronic hepatitis B and HIV. Patent CN112175003B introduces a transformative preparation method for phenyl hydrogen phosphonate and its key intermediates, addressing longstanding bottlenecks in the synthesis of Tenofovir Phenyl Ester. This innovation bypasses the problematic formation of stable tenofovir monohydrate and eliminates the reliance on hazardous reagents like trimethylbromosilane (TMSBr). By utilizing diphenyl hydroxymethylphosphonate as a starting material and employing a controlled low-temperature sulfonylation strategy, this technology offers a scalable, high-purity route that aligns with modern green chemistry principles. For procurement leaders and R&D directors, this represents a significant opportunity to optimize the supply chain for high-purity pharmaceutical intermediates while mitigating regulatory and safety risks associated with traditional synthetic routes.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of Tenofovir Phenyl Ester has been plagued by complex multi-step sequences that hinder industrial scalability and increase production costs. Traditional routes, such as those disclosed by Gilead Sciences, often involve the initial formation of a diethyl ester intermediate followed by a difficult de-ethylation step using expensive and moisture-sensitive reagents like TMSBr. Furthermore, the intermediate tenofovir (PMPA) tends to form a stable monohydrate, necessitating energy-intensive dehydration processes involving azeotropic distillation with cyclohexane or toluene. These steps not only consume significant utility resources but also introduce variability in batch consistency. Additionally, previous attempts to streamline this process, such as those in Chinese patent CN104817593B, failed to reliably produce the necessary activated phosphonate intermediates, often yielding incorrect sulfonate byproducts instead of the target compound. The reliance on toxic solvents like chloroform for extraction further complicates waste management and environmental compliance, making these legacy methods increasingly untenable for modern large-scale manufacturing.
The Novel Approach
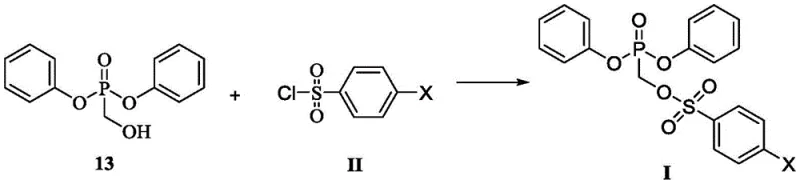
The methodology outlined in CN112175003B fundamentally restructures the synthetic logic by introducing the phenyl ester moiety at the beginning of the sequence rather than at the end. This strategic shift allows for the direct construction of the diphenyl tenofovir ester intermediate (14) without passing through the problematic free acid tenofovir stage. The core innovation lies in the precise preparation of benzenesulfonyloxymethylphosphonate compounds (Formula I) via a low-temperature sulfonylation reaction. By strictly controlling the reaction temperature between -20°C and 0°C, the process achieves high selectivity, avoiding the formation of undesired sulfonate esters that plagued earlier attempts. This approach not only shortens the overall synthetic timeline but also utilizes commercially available and cost-effective raw materials like diphenyl phosphite. The resulting intermediate possesses favorable physicochemical properties, specifically low water solubility, which facilitates straightforward isolation and purification, thereby enhancing the overall throughput and reliability of the manufacturing process for reliable pharmaceutical intermediate supplier operations.
Mechanistic Insights into Low-Temperature Sulfonylation and Coupling
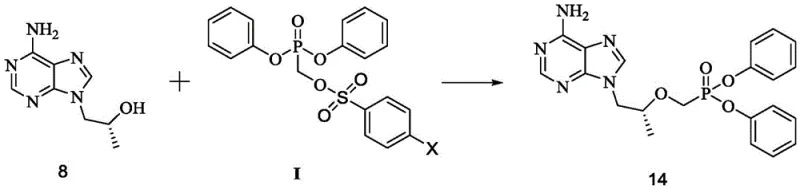
The success of this novel pathway hinges on the mechanistic precision of the sulfonylation step, where diphenyl hydroxymethylphosphonate (13) reacts with a benzenesulfonyl chloride derivative (Formula II). Unlike previous methods that operated at ambient or reflux temperatures leading to side reactions, this patent mandates a cryogenic environment (-20°C to 0°C) in the presence of an acid-binding agent like triethylamine. This thermal control is critical for kinetically favoring the formation of the P-O-C-S linkage over the competing O-S bond formation on the phenyl rings. The subsequent nucleophilic substitution involves a sophisticated activation of the adenine derivative (8) using a Grignard reagent and tert-butanol mixture. This specific combination generates a reactive magnesium alkoxide species in situ, which effectively displaces the benzenesulfonate leaving group on the phosphonate intermediate. This mechanism ensures high regioselectivity for the N9-position of the purine ring, minimizing the formation of N7-isomers and other structural impurities that are difficult to remove in later stages.
Furthermore, the final hydrolysis step demonstrates exceptional chemoselectivity, cleaving one phenyl ester group while preserving the integrity of the rest of the molecule. The use of mild alkaline conditions, such as lithium hydroxide in a THF-water system, allows for the controlled conversion of the diphenyl ester (14) to the target phenyl hydrogen phosphonate (1). This step avoids the harsh acidic conditions often required in other routes, which can lead to depurination or degradation of the sensitive glycosidic bond. The mechanistic robustness of this sequence ensures that the impurity profile remains tightly controlled throughout the synthesis. By avoiding the formation of stable hydrates and utilizing intermediates that are easily crystallized, the process inherently limits the entrapment of solvent residues and metal contaminants. This level of control is paramount for meeting the stringent purity specifications required for active pharmaceutical ingredients, ensuring that the final product is suitable for direct salt formation with fumaric acid to produce TAF.
How to Synthesize Tenofovir Phenyl Ester Efficiently
Implementing this synthesis requires strict adherence to the patented reaction parameters, particularly regarding temperature control and reagent stoichiometry during the sulfonylation phase. The process begins with the preparation of the activated phosphonate species, followed by the coupling with the adenine nucleoside analog, and concludes with a selective hydrolysis. Each step has been optimized to maximize yield while minimizing the generation of hazardous waste. Operators must ensure that the sulfonylation reaction is monitored closely via HPLC or TLC to prevent over-reaction or decomposition, as the window for optimal selectivity is narrow. The work-up procedures leverage standard liquid-liquid extraction techniques but benefit from the favorable partition coefficients of the new intermediates, reducing the need for complex chromatographic separations. For detailed operational protocols and specific safety guidelines regarding the handling of Grignard reagents and sulfonyl chlorides, please refer to the standardized synthesis guide below.
- Perform sulfonylation of diphenyl hydroxymethylphosphonate with benzenesulfonyl chloride at low temperatures (-20 to 0°C) to form the activated phosphonate intermediate.
- Execute nucleophilic substitution using a Grignard reagent and tert-butanol mixture with (R)-9-(2-hydroxypropyl)adenine to couple the phosphonate moiety.
- Conduct alkaline hydrolysis of the resulting diphenyl ester using lithium hydroxide in THF/water to yield the final phenyl hydrogen phosphonate product.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, this patented process offers substantial advantages that directly impact the bottom line and supply chain resilience for manufacturers of antiviral APIs. By eliminating the need for trimethylbromosilane (TMSBr), a reagent known for its high cost, corrosivity, and sensitivity to moisture, the process significantly reduces raw material expenses and storage hazards. The removal of TMSBr also simplifies the engineering controls required for the reactor suite, as there is no longer a need for specialized handling systems for highly corrosive gases or liquids. Furthermore, the avoidance of the tenofovir monohydrate dehydration step translates to reduced energy consumption and shorter cycle times, allowing for faster batch turnover. These efficiencies collectively contribute to significant cost reduction in pharmaceutical intermediate manufacturing, making the final API more competitive in the global market. The use of common solvents like dichloromethane and cyclohexane, rather than restricted solvents like chloroform, further eases regulatory compliance and waste disposal costs.
- Cost Reduction in Manufacturing: The elimination of expensive and specialized reagents like TMSBr and the reduction in processing steps lead to a leaner cost structure. By bypassing the isolation of the free acid tenofovir, the process avoids the yield losses typically associated with multi-step purification and dehydration cycles. The high selectivity of the sulfonylation reaction minimizes the formation of byproducts, reducing the load on downstream purification units and lowering solvent consumption. This streamlined approach ensures that capital expenditure is optimized, as existing equipment can often be retrofitted for this chemistry without requiring exotic materials of construction resistant to extreme corrosion.
- Enhanced Supply Chain Reliability: The reliance on readily available starting materials such as diphenyl phosphite and common sulfonyl chlorides mitigates the risk of supply disruptions often associated with niche reagents. The robustness of the reaction conditions, particularly the tolerance for standard work-up procedures, ensures consistent batch-to-batch quality, which is critical for maintaining long-term supply contracts with major pharmaceutical companies. Additionally, the improved stability of the intermediates allows for more flexible inventory management, reducing the pressure on just-in-time delivery models and providing a buffer against logistical delays. This reliability makes the manufacturer a preferred partner for clients seeking a secure source of high-purity pharmaceutical intermediates.
- Scalability and Environmental Compliance: The process is designed with scalability in mind, utilizing reaction conditions that are easily transferable from pilot plant to commercial scale. The absence of mutagenic solvents like chloroform aligns with increasingly strict environmental regulations and corporate sustainability goals. Waste streams are simpler to treat, as they do not contain complex organophosphorus byproducts or heavy metal residues from catalysts. This environmental friendliness not only reduces the cost of waste treatment but also enhances the company's reputation as a responsible manufacturer. The ability to scale up complex pharmaceutical intermediates efficiently ensures that production can meet surging global demand for antiviral therapies without compromising on quality or safety standards.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this novel synthesis route. These answers are derived directly from the experimental data and technical disclosures within patent CN112175003B, providing clarity on the feasibility and advantages of the method. Understanding these details is crucial for technical teams evaluating the transition from legacy processes to this improved methodology. The insights provided here cover aspects ranging from reaction kinetics to impurity control, ensuring a comprehensive understanding of the technology's value proposition.
Q: Why is the novel sulfonylation temperature critical for yield?
A: Maintaining the reaction temperature between -20°C and 0°C during sulfonylation prevents the formation of side products like phenyl p-toluenesulfonate, ensuring high selectivity for the desired benzenesulfonyloxymethylphosphonate intermediate.
Q: How does this method improve supply chain stability compared to Gilead's route?
A: This process eliminates the need for trimethylbromosilane (TMSBr), a corrosive and moisture-sensitive reagent, and avoids the formation of stable tenofovir monohydrate, significantly simplifying purification and reducing dependency on hazardous solvents like chloroform.
Q: What are the purity advantages of the diphenyl ester intermediate?
A: The diphenyl tenofovir intermediate exhibits low water solubility, allowing for easy extraction and crystallization from aqueous work-up mixtures, which leads to higher purity profiles suitable for downstream API synthesis without extensive chromatography.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Tenofovir Phenyl Ester Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient and compliant manufacturing pathways for life-saving antiviral medications. Our technical team has extensively analyzed the innovations presented in CN112175003B and is fully equipped to implement this advanced synthesis route for the production of Tenofovir Phenyl Ester. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of this patent are realized in practical, large-volume manufacturing. Our facilities are staffed by expert chemists and engineers who specialize in optimizing reaction conditions to achieve stringent purity specifications, utilizing our rigorous QC labs to verify every batch against the highest international standards. We are committed to delivering high-purity pharmaceutical intermediates that empower our partners to bring affordable and effective treatments to patients worldwide.
We invite procurement managers and supply chain directors to engage with us for a Customized Cost-Saving Analysis tailored to your specific production needs. By leveraging this novel synthetic route, we can help you reduce overall manufacturing costs while enhancing supply security. We encourage you to contact our technical procurement team to request specific COA data and route feasibility assessments for your upcoming projects. Together, we can drive innovation in the pharmaceutical supply chain, ensuring the continuous availability of essential medicines through superior chemical manufacturing excellence.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →