Scalable Synthesis of Multi-Target Kinase Inhibitor Intermediates for Oncology Applications
Scalable Synthesis of Multi-Target Kinase Inhibitor Intermediates for Oncology Applications
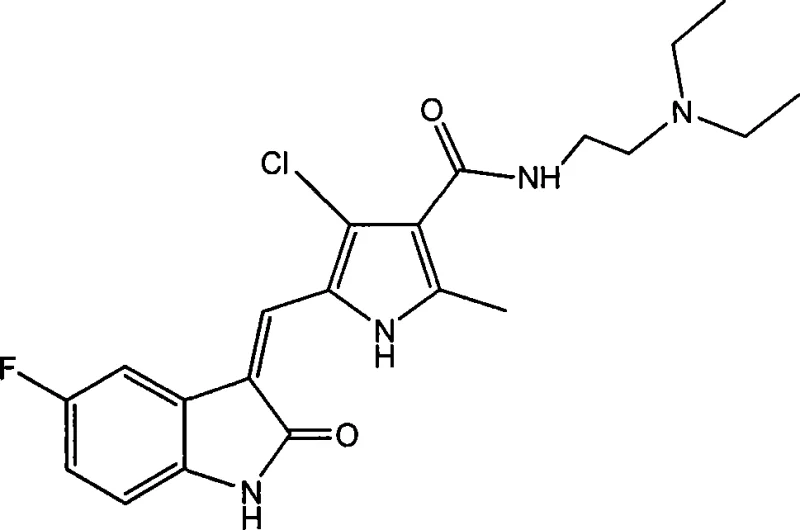
The landscape of oncology drug discovery has shifted dramatically from non-specific cytotoxic agents to targeted therapies that inhibit specific signal transduction pathways. A pivotal development in this arena is the class of multi-target tyrosine kinase inhibitors, exemplified by compounds like Sunitinib. Patent CN101440086B discloses a robust and versatile synthetic methodology for producing halogenated pyrrole substituted 2-indolinones, a core scaffold critical for inhibiting receptors such as VEGFR and PDGFR. For pharmaceutical manufacturers and R&D directors, understanding the nuances of this chemistry is essential for developing next-generation anti-tumor agents with improved efficacy and safety profiles. This report analyzes the technical merits of this patented process, highlighting its potential for cost-effective commercial scale-up and supply chain optimization.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional approaches to synthesizing complex kinase inhibitors often suffer from linear synthetic routes that limit structural diversity and increase production costs. Many legacy processes rely heavily on palladium-catalyzed cross-coupling reactions for constructing the central carbon-carbon bonds. While effective on a small scale, these methods introduce significant challenges during commercial manufacturing, including the removal of trace heavy metals to meet stringent ICH Q3D guidelines. Furthermore, conventional routes often require harsh reaction conditions or protecting group strategies that add unnecessary steps, reducing overall atom economy and increasing waste generation. The inability to easily modify the substitution pattern on the pyrrole or indolinone rings without restarting the synthesis from early intermediates slows down the Structure-Activity Relationship (SAR) exploration phase, delaying the identification of optimal clinical candidates.
The Novel Approach
The methodology described in the patent offers a superior alternative through a convergent synthesis strategy that decouples the construction of the pyrrole and indolinone fragments until the final stages. By utilizing a condensation reaction between a functionalized pyrrole aldehyde and a substituted oxindole, the process allows for independent optimization of each fragment. This modularity is a game-changer for reliable pharmaceutical intermediates supplier networks, as it enables the stocking of diverse building blocks that can be rapidly assembled into various analogues. The use of standard peptide coupling reagents (EDC/HOBt) for attaching the solubilizing side chains avoids the need for exotic catalysts. This approach not only streamlines the workflow but also enhances the purity profile of the final product by minimizing side reactions associated with transition metal catalysis, thereby facilitating easier downstream processing and purification.
Mechanistic Insights into Amide Coupling and Base-Catalyzed Condensation
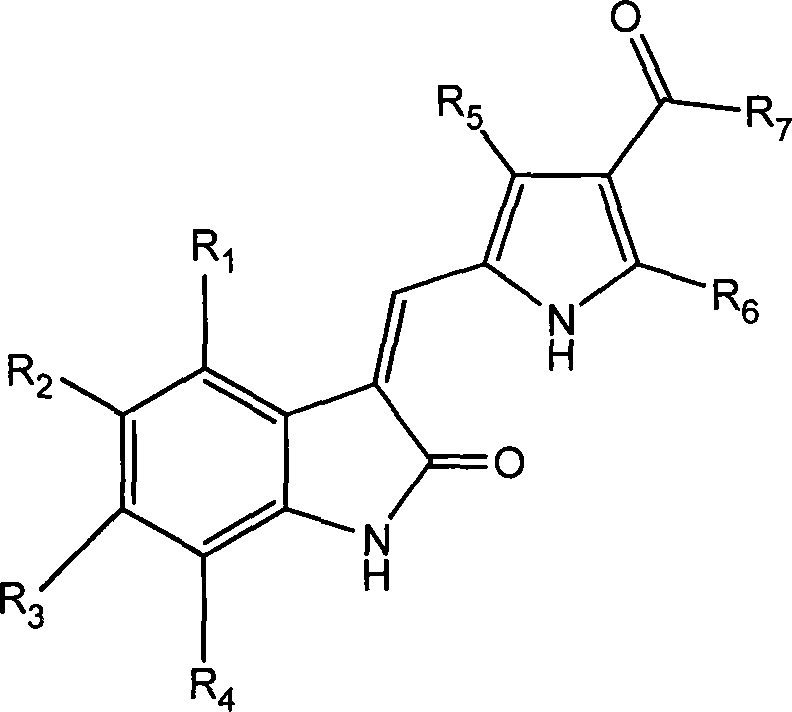
The synthetic pathway hinges on two critical transformations: the formation of the amide bond on the pyrrole ring and the subsequent Knoevenagel-type condensation to link the pyrrole and indolinone systems. The first step involves activating the carboxylic acid moiety of the halogenated pyrrole intermediate (Formula II) using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and 1-hydroxybenzotriazole (HOBt). This activation generates a highly reactive O-acylisourea intermediate, which is stabilized by the HOBt to prevent racemization and rearrangement. The nucleophilic attack by the amine side chain (HR7) proceeds efficiently under mild conditions. Crucially, the patent specifies maintaining the reaction pH between 9 and 14. This alkaline environment ensures the amine nucleophile remains unprotonated and reactive, while also neutralizing the urea byproduct formed during the coupling, driving the equilibrium towards the desired amide product.
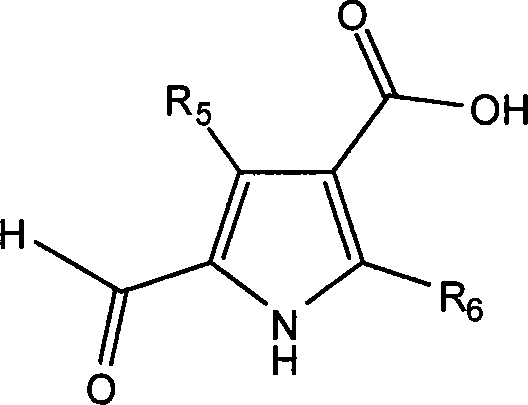
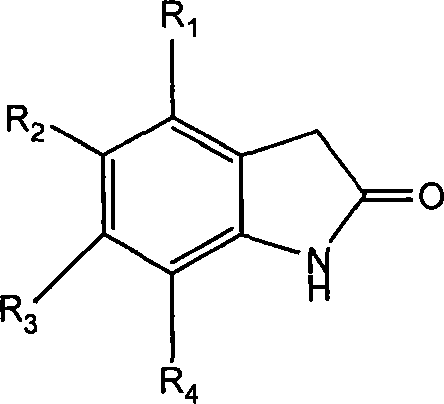
Following the amide formation, the second key step is the condensation with the oxindole derivative (Formula III). This reaction creates the exocyclic double bond connecting the two heterocyclic systems, a structural feature essential for the molecule's ability to bind to the ATP pocket of kinases. The mechanism involves the deprotonation of the methylene group at the 3-position of the oxindole by a base, generating a nucleophilic enolate. This enolate attacks the aldehyde carbonyl of the pyrrole fragment, followed by dehydration to form the stable alkene. The patent recommends conducting this step at elevated temperatures (50-90°C) with a catalytic amount of base. This thermal energy overcomes the activation barrier for the elimination of water, ensuring high conversion rates. The choice of base and solvent system is critical here; using organic bases like triethylamine or pyridine in aprotic or mixed solvent systems helps solubilize the reactants while maintaining the necessary basicity for enolate formation without degrading the sensitive functional groups.
How to Synthesize Halogenated Pyrrole Indolinones Efficiently
Implementing this synthesis requires precise control over reaction parameters to maximize yield and purity. The process begins with the preparation of the key pyrrole acid intermediate, typically via hydrolysis of the corresponding ethyl ester. This step is straightforward, utilizing aqueous base followed by acidification to precipitate the product. Once the acid is secured, the focus shifts to the coupling sequence. Operators must monitor the pH closely during the amide formation to prevent hydrolysis of the activated ester. After isolating the amide intermediate, the final condensation with the oxindole is performed. Workup procedures generally involve extraction into organic solvents like dichloromethane or ethyl acetate, followed by washing to remove water-soluble impurities and salts. The final product can often be purified by recrystallization from ethanol or similar solvents, yielding a high-purity solid suitable for further pharmaceutical development. Detailed standardized synthesis steps are provided in the guide below.
- Prepare the halogenated pyrrole carboxylic acid intermediate via hydrolysis of the corresponding ethyl ester under basic conditions.
- Activate the carboxylic acid using EDC and HOBt, then couple with the desired amine side chain at controlled pH levels.
- Perform the final condensation with a substituted oxindole derivative under basic conditions at elevated temperatures to form the methylene bridge.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, the synthetic route outlined in CN101440086B presents compelling advantages over traditional methods. The reliance on commodity chemicals and the avoidance of precious metal catalysts directly translate to cost reduction in pharmaceutical intermediates manufacturing. By eliminating the need for palladium scavengers and extensive metal testing, manufacturers can significantly lower their operational expenditures and reduce the environmental footprint associated with heavy metal waste disposal. Furthermore, the modularity of the synthesis allows for flexible inventory management; suppliers can stockpile the common pyrrole and oxindone cores and produce specific analogues on demand, reducing lead times for custom synthesis projects.
- Cost Reduction in Manufacturing: The process utilizes widely available reagents such as EDC, HOBt, and common organic bases, which are significantly cheaper than specialized transition metal catalysts. The absence of expensive ligands and the ability to perform reactions in standard glass-lined reactors without special inert atmosphere requirements (beyond standard moisture exclusion) lowers the capital expenditure barrier. Additionally, the high atom economy of the condensation step minimizes raw material waste, contributing to substantial cost savings in large-scale production runs.
- Enhanced Supply Chain Reliability: The starting materials, including substituted oxindoles and pyrrole esters, are commercially available from multiple global vendors, mitigating the risk of single-source supply disruptions. The robust nature of the chemistry means that the process is less sensitive to minor variations in raw material quality, ensuring consistent batch-to-batch reproducibility. This reliability is crucial for maintaining continuous supply lines for clinical trials and eventual commercial launch, preventing costly delays in drug development timelines.
- Scalability and Environmental Compliance: The reaction conditions are mild and scalable, moving seamlessly from gram-scale laboratory synthesis to multi-kilogram pilot plant operations. The use of aqueous workups and the potential for solvent recycling align with green chemistry principles, helping manufacturers meet increasingly strict environmental regulations. The simplified purification process, often requiring only crystallization rather than column chromatography, reduces solvent consumption and waste generation, making the process more sustainable and compliant with modern environmental standards.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the synthesis and application of these compounds. Understanding these details is vital for project managers evaluating the feasibility of incorporating this scaffold into their drug discovery pipelines. The answers are derived directly from the experimental data and claims within the patent documentation, ensuring accuracy and relevance for technical decision-making.
Q: What is the primary advantage of this synthetic route for kinase inhibitors?
A: The route utilizes a convergent strategy that allows for late-stage diversification of both the pyrrole and indolinone moieties, facilitating rapid SAR exploration without redesigning the entire synthesis.
Q: Are heavy metal catalysts required for the final coupling step?
A: No, the final condensation step relies on base catalysis rather than transition metal catalysis, which significantly simplifies purification and reduces the risk of heavy metal contamination in the final API.
Q: What are the typical reaction conditions for the amide formation?
A: The amide coupling is typically performed using EDC/HOBt activation at mild temperatures (0-40°C) with strict pH control between 9 and 14 to ensure high conversion and minimize hydrolysis.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Halogenated Pyrrole Indolinones Supplier
The synthetic pathway detailed in patent CN101440086B represents a significant opportunity for the development of potent multi-target kinase inhibitors. At NINGBO INNO PHARMCHEM, we possess the technical expertise and infrastructure to translate this patented chemistry into commercial reality. Our team has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can move smoothly from preclinical research to market supply. We adhere to stringent purity specifications and operate rigorous QC labs to guarantee that every batch of intermediate meets the highest quality standards required for pharmaceutical applications.
We invite you to collaborate with us to optimize your supply chain for these critical oncology intermediates. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements. Contact us today to request specific COA data and route feasibility assessments, and let us demonstrate how our manufacturing capabilities can support your drug development goals efficiently and reliably.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →