Scalable Production of Sofosbuvir Intermediate VII via Novel Oxidative Deprotection
Scalable Production of Sofosbuvir Intermediate VII via Novel Oxidative Deprotection
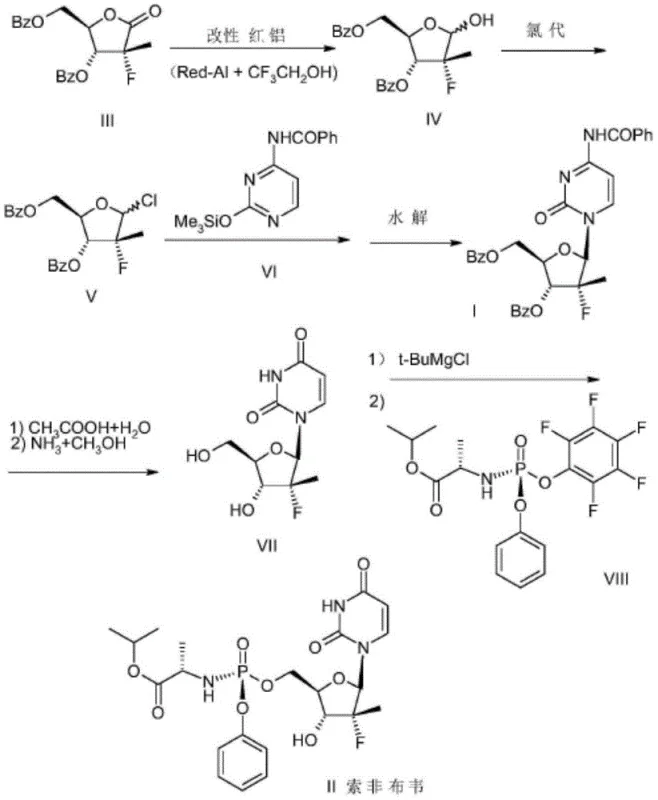
The global demand for direct-acting antiviral agents, particularly for the treatment of chronic Hepatitis C, has placed immense pressure on the supply chains of key pharmaceutical intermediates. Among these, (2'R)-2'-deoxy-2'-fluoro-2'-methyluridine (Compound VII) stands out as a critical building block for the synthesis of Sofosbuvir, a blockbuster drug developed by Gilead Sciences. The efficient manufacturing of this nucleoside analogue is paramount for ensuring the availability of life-saving medications. Recent intellectual property developments, specifically Patent CN107522763B, disclose a groundbreaking preparation method that addresses long-standing inefficiencies in the synthetic route. This patent details a novel two-step transformation starting from the protected cytidine derivative (Compound I), utilizing Lithium Diisopropylamide (LDA) and Sodium Hypochlorite (NaClO) to achieve high purity and yield without the need for cumbersome chromatographic purification.
The significance of this technological advancement cannot be overstated for stakeholders in the fine chemical and pharmaceutical sectors. Traditional methods for synthesizing Compound VII often suffer from low overall yields and reliance on purification techniques that are difficult to scale, such as silica gel column chromatography. These limitations directly impact the cost of goods sold (COGS) and the reliability of supply for downstream API manufacturers. By introducing a streamlined pathway that leverages selective deprotection and oxidative conversion, the inventors have created a process that is not only chemically elegant but also commercially robust. This report provides a deep technical analysis of this novel methodology, evaluating its mechanistic underpinnings and its profound implications for cost reduction and supply chain stability in the production of high-value antiviral intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior to the innovations described in CN107522763B, the industrial standard for converting the protected cytidine precursor (Compound I) into the target uridine intermediate (Compound VII) relied on a harsh and inefficient sequence. As illustrated in the reference literature (e.g., CN101918425), the conventional route typically involves a two-step degradation and deprotection strategy. The first step entails refluxing Compound I in 80% aqueous acetic acid to remove the N-benzoyl protecting group from the cytosine base, yielding an intermediate dibenzoate (Compound IX). While this step achieves a respectable yield of approximately 91%, it subjects the sensitive fluorinated sugar moiety to strongly acidic conditions, which can promote side reactions and epimerization risks.
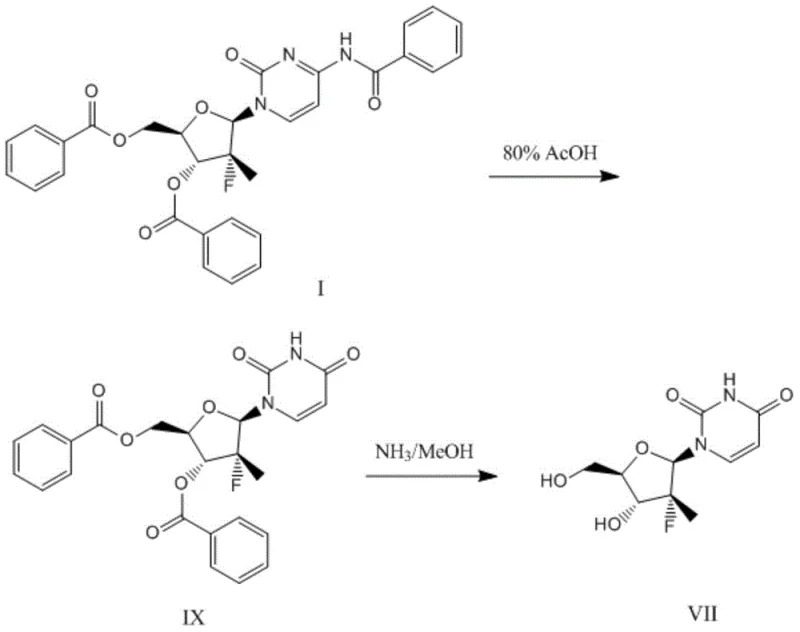
The subsequent step in the traditional process involves treating Compound IX with a saturated solution of ammonia in methanol. This ammonolysis is intended to cleave the benzoate esters on the sugar ring and simultaneously deaminate or hydrolyze the cytosine base to the uracil form. However, this step is notoriously problematic, delivering a poor yield of only about 60%. When combined with the first step, the overall yield for the conversion of Compound I to Compound VII stagnates at roughly 54.6%. Furthermore, the crude product obtained from this sequence is often contaminated with significant impurities, necessitating purification via column chromatography. For large-scale manufacturing, column chromatography is a severe bottleneck; it is labor-intensive, consumes vast amounts of solvents and silica, and limits batch sizes, making it economically unviable for multi-ton production campaigns required by the global Hep C market.
The Novel Approach
In stark contrast to the legacy methods, the novel approach disclosed in Patent CN107522763B reimagines the synthetic logic by decoupling the deprotection and base-modification steps into a highly controlled sequence. Instead of using harsh acid reflux followed by ammonolysis, the new method employs a mild, base-mediated deprotection followed by a selective oxidation. The process begins by treating Compound I with Lithium Diisopropylamide (LDA) in an aprotic solvent like tetrahydrofuran (THF) at low temperatures (0-5°C). This step selectively removes the benzoate protecting groups from the sugar hydroxyls while leaving the N-benzoyl group on the cytosine base intact, yielding Compound X in an impressive 91.7% yield.
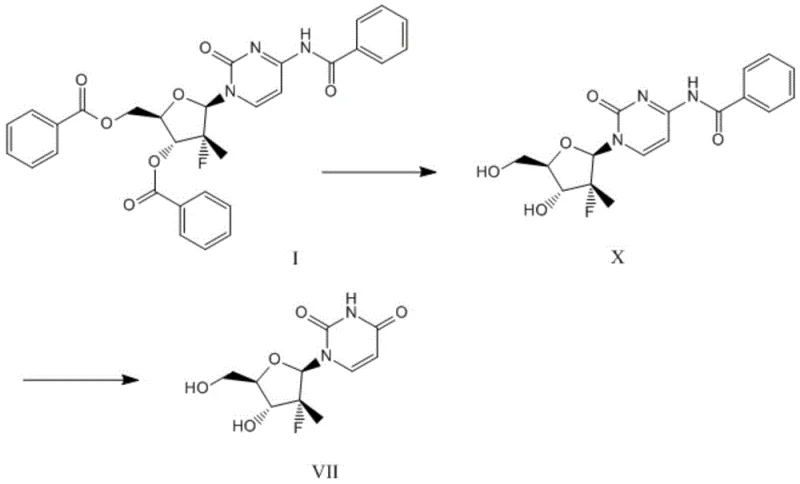
The second transformative step involves the conversion of the cytidine derivative (Compound X) to the uridine target (Compound VII) using Sodium Hypochlorite (NaClO) in the presence of a phase transfer catalyst. This oxidative deamination occurs under mild conditions (20-30°C) and effectively transforms the amino group of the cytosine ring into the carbonyl group of the uracil ring. Crucially, this method avoids the use of toxic ammonia gas or pressurized ammonolysis conditions. The overall yield for this two-step sequence reaches approximately 78%, representing a substantial improvement over the 54.6% benchmark of the prior art. Moreover, the purification strategy shifts entirely from chromatography to crystallization, utilizing acetone and n-heptane, which is a game-changer for industrial scalability and environmental compliance.
Mechanistic Insights into LDA-Mediated Deprotection and Hypochlorite Oxidation
The success of this novel route hinges on the precise chemoselectivity of the reagents employed. In the first step, the use of Lithium Diisopropylamide (LDA) is counter-intuitive yet brilliant. LDA is traditionally known as a strong, non-nucleophilic base used for enolate formation. However, in this specific context involving a fluorinated ribose system, it appears to facilitate the hydrolysis or alcoholysis of the ester bonds at the 3' and 5' positions of the sugar ring. The low temperature control (0-5°C) is critical here; it prevents the base from attacking the amide bond of the N-benzoyl group on the nucleobase or causing elimination reactions on the sensitive 2'-fluoro-2'-methyl sugar scaffold. This selectivity ensures that Compound X is formed with high fidelity, preserving the stereochemical integrity of the chiral centers which is vital for the biological activity of the final API.
The second mechanistic highlight is the oxidative conversion of the cytosine moiety to uracil using Sodium Hypochlorite (NaClO). Mechanistically, this likely proceeds through the chlorination of the exocyclic amine at the C4 position of the pyrimidine ring, forming an N-chloro intermediate. Subsequent hydrolysis of this unstable intermediate leads to the loss of nitrogen (as ammonia or nitrogen gas) and the formation of the C4 carbonyl group characteristic of uracil. The inclusion of a phase transfer catalyst, specifically tetrabutylammonium hydrogen sulfate (Bu4NHSO4), is essential for this transformation. It facilitates the transport of the hypochlorite anion from the aqueous phase into the organic phase where the substrate resides, dramatically increasing the reaction rate and completeness. This oxidative strategy is far cleaner than hydrolytic deamination, which often requires extreme pH or temperature, thereby minimizing the formation of degradation byproducts and simplifying the downstream workup.
How to Synthesize (2'R)-2'-deoxy-2'-fluoro-2'-methyluridine Efficiently
Implementing this novel synthesis route requires strict adherence to the optimized reaction conditions outlined in the patent to ensure maximum yield and purity. The process is designed to be operationally simple, relying on standard unit operations common in fine chemical plants, such as low-temperature dosing, phase separation, and crystallization. The elimination of chromatography means that the process flow is continuous and amenable to automation. Below is the structural framework for the standardized synthesis protocol derived from the patent examples, detailing the critical reagents and isolation techniques necessary for successful execution.
- Deprotect the sugar moiety of Compound I using Lithium Diisopropylamide (LDA) in THF at 0-5°C to yield Compound X.
- Oxidize Compound X using Sodium Hypochlorite (NaClO) with a phase transfer catalyst to convert the cytosine base to uracil, yielding Compound VII.
- Purify the final product via crystallization using acetone and n-heptane, avoiding column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the shift from the conventional AcOH/Ammonia route to the LDA/NaClO methodology represents a significant opportunity for value engineering. The primary driver of value is the drastic improvement in mass efficiency. By boosting the overall yield from roughly 55% to nearly 78%, the new process reduces the consumption of expensive starting materials, such as the protected lactone precursors, by nearly one-third. In the high-volume manufacturing of antiviral APIs, where raw material costs constitute a major portion of the COGS, this yield enhancement translates directly into substantial cost savings per kilogram of the final intermediate. Furthermore, the reduction in waste generation aligns with increasingly stringent environmental regulations, potentially lowering waste disposal fees and improving the sustainability profile of the supply chain.
- Cost Reduction in Manufacturing: The most immediate financial benefit arises from the complete elimination of column chromatography. In traditional processes, chromatography is a hidden cost center, consuming expensive silica gel, large volumes of high-purity solvents, and significant labor hours for packing and running columns. By replacing this with a simple crystallization using acetone and n-heptane, the new method drastically cuts processing time and solvent costs. Additionally, the higher yield means less raw material is wasted, further driving down the unit cost of production. This lean manufacturing approach allows suppliers to offer more competitive pricing for Sofosbuvir intermediates without compromising margins.
- Enhanced Supply Chain Reliability: Supply continuity is often threatened by complex purification steps that are prone to variability and bottlenecks. Chromatography is notoriously difficult to scale; a process that works on a 100-gram lab scale often fails or becomes prohibitively slow at the 100-kilogram or ton scale. The new crystallization-based purification is inherently scalable and robust, ensuring that production timelines are met consistently. The use of common, commodity reagents like LDA (or its precursors), NaClO (bleach), and standard solvents like THF and heptane ensures that the supply chain is not dependent on exotic or scarce catalysts, mitigating the risk of raw material shortages.
- Scalability and Environmental Compliance: From an EHS (Environment, Health, and Safety) perspective, the new route offers distinct advantages. The avoidance of refluxing acetic acid and the handling of saturated ammonia solutions reduces the risk of operator exposure to corrosive and toxic vapors. Sodium hypochlorite is a widely available and relatively safe oxidant compared to heavy metal oxidants. Moreover, the solvent system (THF/Acetone/Heptane) is easier to recover and recycle than the complex mixtures often generated in chromatographic elutions. This facilitates a greener manufacturing process with a lower carbon footprint, which is increasingly a requirement for Tier 1 pharmaceutical suppliers aiming to meet corporate sustainability goals.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this novel synthesis route. These insights are derived directly from the experimental data and comparative analysis provided in Patent CN107522763B. Understanding these nuances is critical for R&D teams evaluating technology transfer and for quality assurance teams establishing specification limits for the new intermediate.
Q: Why is the new LDA/NaClO route superior to the traditional AcOH/Ammonia method?
A: The new route eliminates the need for column chromatography, which is a major bottleneck in industrial scale-up. It also improves the overall yield from approximately 54.6% to 78%, significantly reducing raw material costs.
Q: What are the critical process parameters for the oxidation step?
A: The oxidation using NaClO requires precise temperature control between 20-30°C and the use of a phase transfer catalyst like Bu4NHSO4 to ensure efficient conversion of the cytosine derivative to the uridine product.
Q: Is this process suitable for multi-ton production?
A: Yes, the replacement of chromatographic purification with crystallization (using acetone/n-heptane) makes this process highly scalable and compliant with GMP standards for commercial API manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Sofosbuvir Intermediate Supplier
The technological breakthroughs detailed in Patent CN107522763B underscore the dynamic nature of nucleoside analogue manufacturing, where continuous process improvement is key to maintaining competitiveness. At NINGBO INNO PHARMCHEM, we pride ourselves on staying at the forefront of these developments, leveraging advanced synthetic methodologies to deliver superior value to our global partners. Our facility is equipped with the specialized infrastructure required to handle sensitive organometallic reagents like LDA and oxidative transformations safely and efficiently. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that our clients receive a consistent supply of high-quality intermediates regardless of market demand fluctuations.
We understand that transitioning to a new synthetic route requires confidence in both the chemistry and the partner executing it. Our rigorous QC labs employ state-of-the-art analytical instrumentation to verify that every batch meets stringent purity specifications, fully compliant with cGMP standards. We invite procurement leaders and technical directors to engage with us for a Customized Cost-Saving Analysis. By reviewing your current supply chain challenges, our technical procurement team can provide specific COA data and route feasibility assessments to demonstrate how our optimized manufacturing capabilities can enhance your bottom line.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →