Scalable Synthesis of Ganciclovir Mono-Valine Ester via Controlled Hydrolysis
Scalable Synthesis of Ganciclovir Mono-Valine Ester via Controlled Hydrolysis
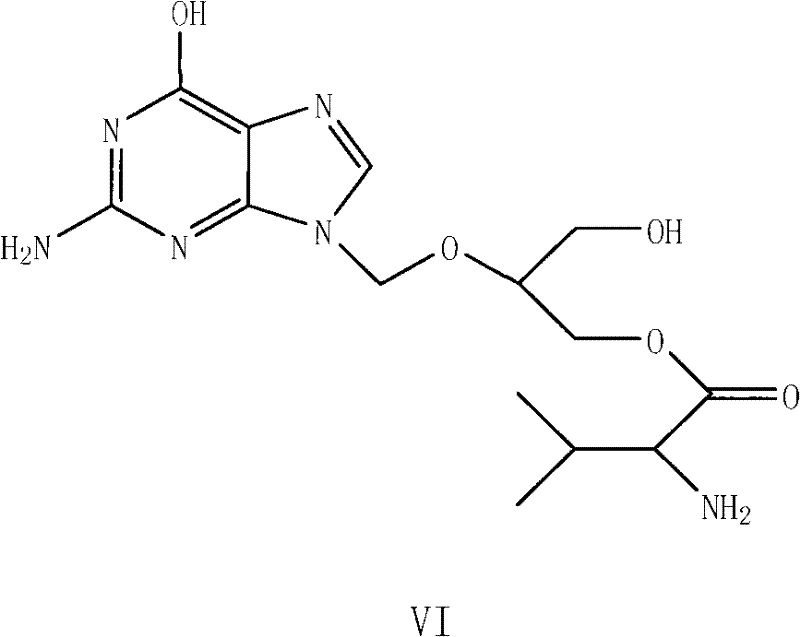
The pharmaceutical industry continuously seeks robust, scalable pathways for producing high-value antiviral intermediates, particularly those serving as precursors to blockbuster drugs like Valganciclovir. Patent CN102070635A introduces a transformative preparation method for ganciclovir valine ester derivatives, specifically targeting the synthesis of ganciclovir-CBZ-L-mono-valine ester. This intermediate is pivotal in the manufacturing chain, acting as the direct precursor to the active prodrug. The disclosed technology represents a significant leap forward in process chemistry, moving away from laborious purification techniques toward a streamlined, kinetically controlled hydrolysis strategy. By utilizing ganciclovir-CBZ-L-di-valine ester as the starting material and employing precise basic catalysis, the method achieves exceptional purity levels exceeding 99.0% without the need for expensive and time-consuming column chromatography. This innovation addresses critical bottlenecks in pharmaceutical intermediates manufacturing, offering a route that is not only chemically elegant but also commercially viable for multi-ton production scales.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art methodologies for synthesizing valganciclovir intermediates have historically been plagued by significant inefficiencies and environmental concerns. For instance, World Patent WO9727195 describes a route involving the selective removal of an ester group from a diester; however, this process relies heavily on organic amine catalysts which possess strong pungency and pose detrimental effects on the working environment and operator safety. Furthermore, the selectivity of these conventional hydrolysis reactions is often poor, resulting in a crude single L-valine ester content of merely 60-70%. Such low purity necessitates complex downstream processing, often involving multiple solvent systems for extraction and separation to distinguish between monoesters, diesters, and free ganciclovir. The reliance on trifluoroacetic acid in some variations introduces additional corrosion issues and waste disposal challenges. These factors collectively render traditional methods unsuitable for modern, green, and cost-effective industrial production, creating a pressing need for a more refined synthetic strategy.
The Novel Approach
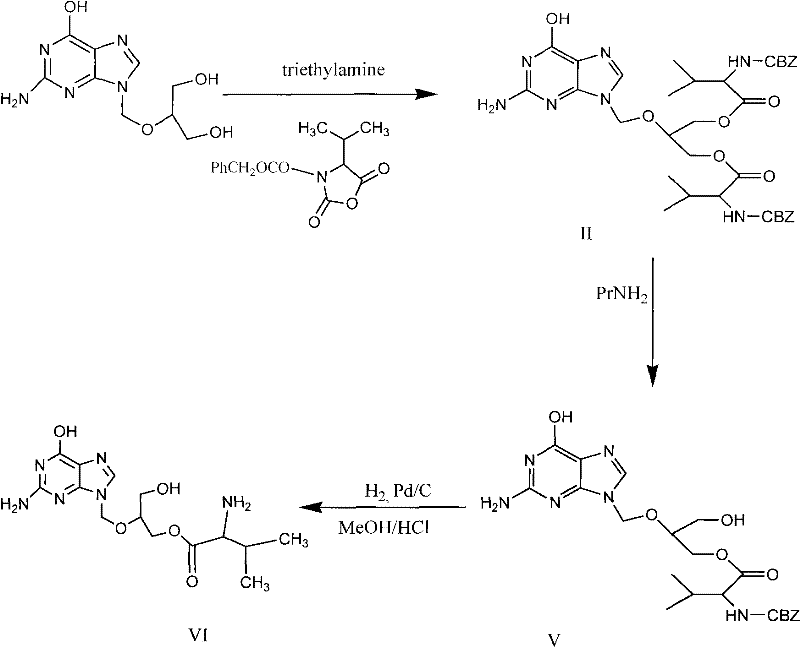
The methodology outlined in CN102070635A fundamentally reengineers the synthesis by optimizing the hydrolysis of the diester precursor. Instead of struggling with low selectivity, this novel approach leverages precise reaction monitoring to halt the process at the exact moment of maximum mono-ester formation. The process involves dissolving the ganciclovir-CBZ-L-di-valine ester in a tailored solvent system—such as a mixture of acetone and methanol—and introducing a mild inorganic base like sodium hydroxide or potassium carbonate. The reaction is maintained at a controlled temperature range of 40-50°C, ensuring kinetic stability. Crucially, the reaction is tracked via Thin Layer Chromatography (TLC) and stopped when the conversion of the diester reaches approximately 45-55%, ideally near 50%. This kinetic control prevents the over-hydrolysis of the desired mono-ester back into ganciclovir. Following the reaction, a sophisticated workup involving pH-adjusted extractions and selective crystallization yields a product with purity surpassing 99.0%, effectively bypassing the need for chromatographic purification entirely.
Mechanistic Insights into Base-Catalyzed Selective Hydrolysis
The core chemical transformation driving this process is a nucleophilic acyl substitution, specifically the base-catalyzed hydrolysis of an ester bond. In the presence of a hydroxide ion (from NaOH or KOH), the nucleophile attacks the carbonyl carbon of the valine ester moiety on the ganciclovir scaffold. This forms a tetrahedral intermediate which subsequently collapses to release the valine carboxylate and the alcohol group on the ganciclovir side chain. The challenge in this system lies in the presence of two identical ester groups on the diester starting material. Statistically, the hydrolysis of the first ester is faster than the second due to steric and electronic factors, but without control, the reaction proceeds to the fully hydrolyzed ganciclovir. The innovation here is the recognition that the concentration of the mono-ester species peaks when roughly half of the diester has reacted. By quenching the reaction at this specific conversion point (around 50%), the process maximizes the yield of the intermediate species before it degrades further. This requires precise analytical monitoring, typically achieved through TLC, comparing the spot intensity of the mono-ester against the remaining di-ester.
Impurity control is managed through a combination of pH manipulation and solubility differences during the workup phase. After the hydrolysis is complete, the reaction mixture contains the desired mono-ester, unreacted di-ester, and potentially some over-hydrolyzed ganciclovir. The process employs a liquid-liquid extraction strategy where the pH is adjusted to specific ranges (e.g., pH 2-7) to selectively partition these components between aqueous and organic phases. The mono-ester, possessing both acidic (carboxylic acid after acidification) and basic (amine) functionalities, exhibits unique solubility profiles compared to the more lipophilic di-ester and the more hydrophilic ganciclovir. By carefully regulating the pH during the extraction and subsequent precipitation steps, impurities are left in the mother liquor while the target compound crystallizes out. This physicochemical purification is far more scalable and robust than adsorption-based methods like column chromatography, ensuring that the final product meets stringent pharmaceutical specifications with minimal residual impurities.
How to Synthesize Ganciclovir-CBZ-L-mono-valine Ester Efficiently
The synthesis of this critical antiviral intermediate requires strict adherence to the optimized parameters defined in the patent to ensure reproducibility and high yield. The process begins with the dissolution of the diester starting material in a polar aprotic or protic solvent system, followed by the controlled addition of a basic catalyst. Operators must maintain the reaction temperature within the narrow window of 40-50°C to balance reaction rate against selectivity loss. Continuous monitoring of the reaction progress is mandatory; the reaction must be quenched immediately once the TLC indicates that the mono-ester spot intensity is comparable to, but not greater than, the di-ester spot. Following quenching, the workup involves a series of extractions using immiscible organic solvents like dichloromethane and acidic aqueous solutions to separate the product from the unreacted starting material. The final purification step utilizes recrystallization from solvents such as methanol or ethanol, potentially with activated carbon treatment for decolorization, to deliver the high-purity solid. For a detailed, step-by-step standard operating procedure including exact stoichiometric ratios and safety protocols, please refer to the technical guide below.
- Dissolve ganciclovir-CBZ-L-di-valine ester in a solvent system such as acetone/methanol and add a basic catalyst like sodium hydroxide at 40-50°C.
- Monitor the reaction via TLC until the di-ester conversion reaches approximately 50% to maximize mono-ester selectivity.
- Perform acid-base extraction to isolate the crude product, followed by recrystallization to achieve purity greater than 99.0% without column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the transition to this novel hydrolysis method offers substantial strategic benefits, primarily driven by the simplification of the manufacturing workflow. Traditional routes that rely on column chromatography are inherently batch-limited, solvent-intensive, and difficult to scale beyond pilot plant quantities. By eliminating the chromatography step entirely, this new process drastically reduces the consumption of silica gel and vast volumes of elution solvents, which are major cost drivers in fine chemical production. Furthermore, the use of common inorganic bases like sodium hydroxide or sodium carbonate replaces expensive and hazardous organic amines, leading to significant raw material cost reductions. The simplified workup, which relies on filtration and crystallization rather than complex distillation or chromatographic separation, shortens the overall cycle time per batch. This efficiency translates directly into improved throughput and lower operational expenditures, making the cost reduction in pharmaceutical intermediates manufacturing a tangible reality rather than just a theoretical projection.
- Cost Reduction in Manufacturing: The elimination of column chromatography is the single most impactful economic driver of this technology. Chromatographic purification requires specialized equipment, large quantities of stationary phase (silica), and massive volumes of organic solvents that must be recovered or disposed of, all of which inflate the cost of goods sold (COGS). By shifting to a crystallization-based purification, the process leverages standard stainless steel reactors and filtration units that are ubiquitous in chemical plants. Additionally, the replacement of corrosive reagents like trifluoroacetic acid with mild mineral acids and bases reduces equipment maintenance costs and extends the lifespan of reactor vessels. The high molar yield (>50%) relative to the starting diester ensures that raw material utilization is optimized, minimizing waste and maximizing the value extracted from every kilogram of input material.
- Enhanced Supply Chain Reliability: Supply continuity is often threatened by the reliance on specialized reagents or complex purification services. This method utilizes commodity chemicals—acetone, methanol, sodium hydroxide, and hydrochloric acid—which are readily available in the global market with stable pricing and secure supply lines. The robustness of the crystallization process means that production is less susceptible to the variability often seen in chromatographic separations, where column packing inconsistencies can lead to batch failures. This reliability allows for more accurate demand forecasting and inventory planning. Moreover, the ability to recycle unreacted di-ester (which can be recovered from the organic phase and reused in subsequent batches) further secures the supply chain by reducing the net consumption of the valuable starting material, thereby insulating production schedules from raw material shortages.
- Scalability and Environmental Compliance: Scaling a chemical process from grams to tons often exposes hidden flaws, but this hydrolysis route is designed with scalability in mind. The unit operations involved—dissolution, heated reaction, liquid-liquid extraction, and crystallization—are standard chemical engineering processes that scale linearly without significant loss of efficiency. From an environmental perspective, the process generates significantly less hazardous waste compared to prior art. The avoidance of pungent organic amines improves workplace safety and reduces the burden on废气 (waste gas) treatment systems. The solvents used are largely recyclable, and the absence of silica gel waste simplifies solid waste disposal. This alignment with green chemistry principles not only reduces disposal costs but also ensures compliance with increasingly stringent environmental regulations, safeguarding the facility against regulatory risks and enhancing the corporate sustainability profile.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production of ganciclovir valine ester derivatives. These answers are derived directly from the experimental data and process descriptions found in the patent literature, providing a transparent view of the technology's capabilities. Understanding these details is essential for technical teams evaluating the feasibility of integrating this route into their existing manufacturing portfolios. The focus is on clarity regarding reaction control, purity assurance, and the practical advantages over legacy methods.
Q: Why is the reaction conversion limited to approximately 50%?
A: Limiting conversion to around 50% is critical for kinetic selectivity. Continuing the reaction beyond this point leads to the over-hydrolysis of the desired mono-ester into ganciclovir, significantly reducing yield. Stopping at the equilibrium point where mono-ester concentration peaks ensures optimal recovery.
Q: How does this method achieve high purity without column chromatography?
A: The process utilizes a sophisticated pH-controlled extraction sequence followed by selective crystallization. By adjusting the pH to precipitate the product while keeping impurities in solution, and subsequently recrystallizing from specific solvent systems like DMF/water or methanol, purity exceeding 99.0% is achieved industrially.
Q: What are the advantages over prior art methods like WO9727195?
A: Unlike previous methods that relied on pungent organic amines and yielded products with only 60-70% purity requiring complex purification, this novel approach uses mild inorganic bases and achieves over 99% purity directly through crystallization, making it far more suitable for large-scale industrial production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Ganciclovir-CBZ-L-mono-valine Ester Supplier
At NINGBO INNO PHARMCHEM, we recognize that the successful commercialization of antiviral therapeutics depends on the availability of high-quality, consistently supplied intermediates. Our technical team has thoroughly analyzed the hydrolysis pathway described in CN102070635A and possesses the extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production required to bring this process to life. We understand the nuances of kinetic control in ester hydrolysis and the critical importance of crystallization parameters in achieving the requisite stringent purity specifications demanded by global regulatory bodies. Our rigorous QC labs are equipped to monitor reaction progress via HPLC and TLC with precision, ensuring that every batch meets the >99.0% purity benchmark without the need for chromatographic intervention. We are committed to delivering this complex intermediate with the reliability and quality that top-tier pharmaceutical manufacturers expect.
We invite procurement leaders and R&D directors to engage with us to explore how this optimized synthesis can enhance your supply chain resilience. By partnering with us, you gain access to a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating exactly how the elimination of chromatography impacts your bottom line. We encourage you to contact our technical procurement team to request specific COA data from our pilot runs and comprehensive route feasibility assessments. Let us collaborate to streamline your production of valganciclovir precursors, ensuring a steady flow of high-purity materials that keep your drug development pipelines moving forward efficiently and economically.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →