Advanced Synthesis of Quinoline and Quinazoline PFKFB3 Inhibitors for Oncology Applications
Introduction to Patent CN109134434B and Glycolysis Inhibition
The landscape of oncology drug development is increasingly focused on metabolic reprogramming, specifically the Warburg effect, where tumor cells rely heavily on glycolysis for energy. Patent CN109134434B introduces a novel class of quinoline and quinazoline compounds designed to disrupt this metabolic pathway by inhibiting the bifunctional enzyme PFKFB3. This technical disclosure provides a comprehensive preparation method that allows for significant structural diversity, enabling the fine-tuning of pharmacokinetic properties while maintaining potent enzymatic inhibition. The disclosed synthesis routes are characterized by their modularity and reliance on accessible chemical building blocks, making them highly attractive for industrial translation. By targeting the energy supply of tumor cells at the source, these compounds offer a promising therapeutic strategy that differs from traditional cytotoxic agents. The following analysis details the chemical ingenuity behind these structures and their potential impact on the supply of high-purity pharmaceutical intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional approaches to synthesizing heterocyclic kinase inhibitors often suffer from harsh reaction conditions, limited substrate scope, and the requirement for expensive transition metal catalysts that are difficult to remove to trace levels. Many existing routes for quinoline derivatives involve high-temperature cyclizations that can lead to significant decomposition or the formation of difficult-to-separate regioisomers. Furthermore, conventional methods for introducing diverse side chains at the 4-position of the quinoline ring frequently require multiple protection and deprotection steps, increasing the overall step count and reducing the cumulative yield. The reliance on specialized reagents can also create bottlenecks in the supply chain, leading to inconsistent availability and inflated costs for research and development teams. These factors collectively hinder the rapid optimization of lead compounds and delay the transition from bench-scale discovery to commercial manufacturing.
The Novel Approach
The methodology outlined in the patent overcomes these hurdles through a streamlined, convergent synthesis that prioritizes operational simplicity and chemical efficiency. By utilizing a diphenyl ether intermediate formed via nucleophilic aromatic substitution, the process establishes the core carbon framework early, allowing for subsequent diversification. The cyclization steps, whether forming the quinoline core via condensation with beta-keto esters or the quinazoline core via reaction with chloral hydrate and urea, proceed under controlled conditions that minimize side reactions. A key advantage is the late-stage functionalization capability, where the nitro group is reduced to an amine and subsequently coupled with a wide array of carboxylic acids. This modularity enables the rapid generation of analog libraries without redesigning the entire synthetic route, significantly accelerating the structure-activity relationship (SAR) studies required for drug candidate selection.
Mechanistic Insights into PFKFB3 Inhibition and Scaffold Construction
The biological efficacy of these compounds stems from their ability to bind to the kinase domain of PFKFB3, preventing the phosphorylation of fructose-6-phosphate to fructose-2,6-biphosphate. Since Fru-2,6-BP is the most potent allosteric activator of phosphofructokinase-1 (PFK-1), its depletion effectively shuts down the glycolytic flux. Chemically, the construction of the pharmacophore relies on precise control over the electronic properties of the heterocyclic ring. The initial formation of the diphenyl ether linkage is critical, as it positions the electron-withdrawing nitro group ortho to the phenolic oxygen, facilitating the subsequent intramolecular cyclization. The use of polyphosphoric acid (PPA) in the quinoline synthesis acts as both a solvent and a dehydrating agent, promoting the condensation of the aniline derivative with the beta-keto ester to form the 4-hydroxyquinoline intermediate. This intermediate is then activated via O-alkylation, creating a leaving group that can be displaced by nucleophiles or simply serving as a stable ether linkage depending on the specific embodiment.
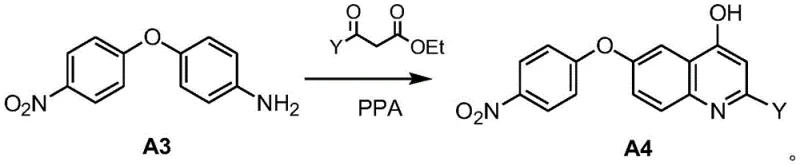
Impurity control is managed through the selection of reagents that favor thermodynamic products and the implementation of robust workup procedures. For instance, the reduction of the nitro group using zinc powder and ammonium chloride is highly chemoselective, avoiding the reduction of other sensitive functional groups that might be present on the side chains. The final amide coupling step utilizes carbodiimide chemistry, such as EDCI, which is well-understood and allows for the efficient removal of urea byproducts through aqueous extraction. The structural rigidity provided by the fused ring system ensures that the molecule maintains the correct conformation for enzyme binding, while the variable R groups allow for optimization of solubility and metabolic stability. This balance between structural integrity and chemical flexibility is what makes this scaffold particularly valuable for developing next-generation antineoplastic agents.
How to Synthesize Quinoline or Quinazoline Compounds Efficiently
The synthesis of these potent PFKFB3 inhibitors follows a logical sequence of transformations that can be adapted for both small-scale medicinal chemistry and large-scale process development. The route begins with the protection of 4-aminophenol, followed by etherification to install the nitrophenyl group, setting the stage for ring closure. Detailed standardized synthetic steps for the preparation of the core intermediates and final compounds are provided in the guide below, ensuring reproducibility and consistency across different batches. The process emphasizes the use of common organic solvents and reagents, minimizing the need for specialized equipment or hazardous conditions. Operators should pay close attention to the stoichiometry during the cyclization and coupling steps to maximize yield and minimize the formation of oligomeric byproducts.
- Perform nucleophilic substitution on protected aminophenol with 4-fluoronitrobenzene to form the diphenyl ether backbone.
- Execute condensation cyclization using polyphosphoric acid or chloral hydrate to construct the quinoline or quinazoline core scaffold.
- Reduce the nitro group to an amine and perform final amide coupling with diverse carboxylic acids to generate the target inhibitor.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement perspective, the synthetic route described in the patent offers substantial advantages in terms of raw material availability and cost structure. The starting materials, including 4-aminophenol, Boc anhydride, and various halo-nitrobenzenes, are commodity chemicals produced in high volumes globally, ensuring a stable supply base that is not subject to the volatility often seen with exotic reagents. The elimination of precious metal catalysts, such as palladium or platinum, which are common in cross-coupling reactions, removes a significant cost driver and simplifies the purification process. This reduction in material complexity translates directly into lower manufacturing costs and a more predictable pricing model for long-term supply agreements. Additionally, the avoidance of cryogenic conditions or high-pressure hydrogenation equipment reduces the capital expenditure required for production facilities.
- Cost Reduction in Manufacturing: The process leverages inexpensive reagents like zinc powder for reduction and polyphosphoric acid for cyclization, which are significantly cheaper than catalytic hydrogenation systems or specialized Lewis acids. By avoiding the use of transition metals, the downstream processing is simplified, as there is no need for expensive metal scavenging resins or rigorous testing for residual heavy metals, which are critical quality attributes for pharmaceutical ingredients. The high atom economy of the condensation reactions further contributes to waste reduction, lowering the environmental disposal costs associated with production. Overall, the streamlined nature of the synthesis allows for a more efficient allocation of resources, driving down the cost of goods sold (COGS) for the final active pharmaceutical ingredient.
- Enhanced Supply Chain Reliability: The reliance on widely available bulk chemicals mitigates the risk of supply disruptions that can occur with single-source specialty reagents. The synthetic steps are robust and tolerant of minor variations in reaction parameters, which enhances the reliability of the manufacturing process and ensures consistent on-time delivery. The modular design of the synthesis means that if a specific side-chain acid becomes unavailable, alternative analogs can be synthesized with minimal process changes, providing flexibility in the face of market fluctuations. This resilience is crucial for maintaining continuity of supply for clinical trials and eventual commercial launch, safeguarding against potential bottlenecks in the global chemical supply chain.
- Scalability and Environmental Compliance: The reaction conditions are mild, typically ranging from room temperature to moderate heating, which facilitates safe scale-up from kilogram to multi-ton quantities without significant engineering challenges. The use of standard solvents like dichloromethane, methanol, and ethyl acetate allows for established recovery and recycling protocols, aligning with green chemistry principles and regulatory environmental standards. The absence of highly toxic or explosive reagents reduces the safety risks associated with large-scale operations, lowering insurance premiums and regulatory compliance burdens. This combination of safety, scalability, and environmental stewardship makes the technology highly attractive for contract development and manufacturing organizations (CDMOs) looking to optimize their production portfolios.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these quinoline and quinazoline derivatives. The answers are derived from the detailed experimental data and mechanistic explanations provided in the patent documentation, offering clarity on the feasibility and benefits of this technology. Understanding these aspects is essential for stakeholders evaluating the potential integration of these compounds into their drug development pipelines. The information provided here serves as a foundational guide for further technical discussions and partnership opportunities.
Q: What is the primary mechanism of action for these quinoline compounds?
A: These compounds function as PFKFB3 inhibitors, blocking the synthesis of Fructose-2,6-biphosphate (Fru-2,6-BP). This effectively inhibits the rate-limiting enzyme PFK-1 in the glycolytic pathway, starving tumor cells of energy.
Q: Are the raw materials for this synthesis commercially available?
A: Yes, the synthesis utilizes readily available starting materials such as 4-aminophenol, Boc anhydride, and various substituted ethyl acetoacetates, ensuring a robust and reliable supply chain for manufacturing.
Q: How does this process address impurity control during scale-up?
A: The process employs mild reaction conditions and standard purification techniques like silica gel column chromatography and recrystallization, which are highly effective at removing side products and ensuring high purity specifications suitable for pharmaceutical applications.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Quinoline Compound Supplier
The technological potential of these PFKFB3 inhibitors represents a significant opportunity for advancing oncology therapeutics, and NINGBO INNO PHARMCHEM is uniquely positioned to support this journey. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can transition smoothly from preclinical research to market supply. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications required for global regulatory filings. We understand the critical nature of timeline and quality in drug development and are committed to delivering high-performance intermediates that meet the exacting standards of the pharmaceutical industry.
We invite you to engage with our technical procurement team to discuss how we can optimize your supply chain for these complex heterocyclic compounds. By requesting a Customized Cost-Saving Analysis, you can gain insights into potential efficiencies and cost reductions specific to your volume requirements. We encourage you to reach out for specific COA data and route feasibility assessments to validate the compatibility of our manufacturing capabilities with your project needs. Let us collaborate to bring these innovative glycolysis inhibitors to patients faster and more efficiently.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →