Advanced Synthesis of Cefminox Impurity D for Regulatory Compliance and Quality Control
Advanced Synthesis of Cefminox Impurity D for Regulatory Compliance and Quality Control
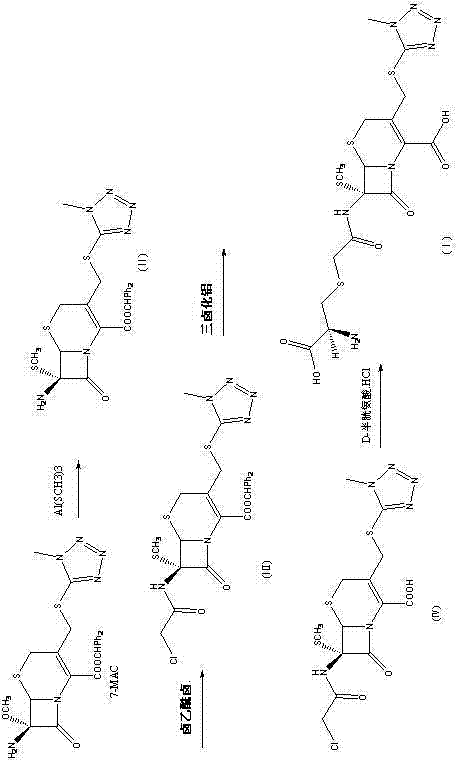
The pharmaceutical industry faces rigorous challenges in maintaining the quality and safety of antibiotic formulations, particularly for complex cephamycins like Cefminox Sodium. A critical aspect of this quality assurance is the identification and quantification of process-related impurities, which can arise from subtle variations in synthetic pathways. Patent CN102875577B addresses a significant gap in this domain by disclosing a robust, multi-step preparation method for Cefminox Impurity D, a specific degradation product associated with the methoxylation of the key intermediate 7-MAC. Unlike previous methods that relied on difficult isolation from crude reaction mixtures—often resulting in poor purity levels below 80%—this invention provides a deliberate synthetic route to generate high-purity reference standards. For R&D directors and quality control managers, access to such well-characterized impurities is not merely a regulatory checkbox but a fundamental requirement for validating analytical methods and ensuring the stability of the final drug product.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the procurement of specific antibiotic impurities like Impurity D has been fraught with technical difficulties and supply chain inconsistencies. In traditional manufacturing scenarios, these impurities were often unintended byproducts formed during the large-scale synthesis of the active pharmaceutical ingredient (API). Attempting to isolate them from the crude reaction mass involves complex preparative liquid chromatography, which is both time-consuming and inefficient. Furthermore, beta-lactam antibiotics are inherently unstable; they are prone to hydrolysis and thermal degradation, meaning that during the prolonged isolation processes required to extract trace impurities, the target molecule often decomposes. This results in reference materials that are chemically ill-defined, with purities frequently failing to meet the stringent requirements of pharmacopoeias. Consequently, pharmaceutical companies struggle to validate their HPLC methods accurately, leading to potential delays in regulatory filings and increased risk of batch rejections due to unquantified related substances.
The Novel Approach
The methodology outlined in patent CN102875577B represents a paradigm shift from passive isolation to active, controlled synthesis. Instead of hoping to find the impurity in a waste stream, this approach constructs the molecule deliberately from commercially available starting materials. By utilizing 7-MAC (7-methoxycephalosporanic acid derivative) as the core scaffold and employing specific reagents like aluminum mercapto and D-cysteine hydrochloride, the process ensures that the structural integrity of the impurity matches exactly what is found in the degraded API. This targeted synthesis allows for the optimization of each reaction step to maximize yield and minimize side products. The result is a reference standard with purity levels capable of exceeding 97%, providing a solid foundation for analytical validation. This shift not only secures the supply of critical reference materials but also offers deep mechanistic insights into how the impurity forms, enabling process chemists to tweak the main API synthesis to suppress its formation in the first place.
Mechanistic Insights into Aluminum Mercapto Mediated Substitution
The core chemical innovation in this patent lies in the use of aluminum mercapto species to facilitate the introduction of the methylthio group, mimicking the side reaction that occurs during the industrial production of Cefminox. The mechanism begins with the generation of an aluminum mercapto complex by reacting aluminum trihalide (such as AlCl3) with methyl mercaptan in a solvent like dichloromethane. This Lewis acid-base complex activates the sulfur nucleophile, making it sufficiently reactive to displace the leaving group at the 3-position of the cephem nucleus under mild conditions. This step is crucial because it avoids the harsh conditions that might otherwise open the sensitive beta-lactam ring. Following this substitution, the intermediate undergoes acylation with a haloacetyl halide, introducing the necessary side chain precursor. The subsequent deprotection step, utilizing trifluoroacetic acid or aluminum trihalide in the presence of a Lewis base like anisole, carefully removes protecting groups without compromising the stereochemistry at the chiral centers. Finally, the coupling with D-cysteine hydrochloride completes the structure, forming the amide bond that characterizes Impurity D. Each step is optimized for temperature and pH, typically keeping reactions between -10°C and 20°C to preserve the fragile bicyclic system.
From an impurity control perspective, understanding this mechanism is vital for root cause analysis. The patent elucidates that Impurity D arises when the 7-amino protecting group falls off during the methoxylation route, releasing methyl mercaptan which then participates in a side reaction. By replicating this pathway in a controlled environment, manufacturers can generate authentic samples to spike into their analytical runs. This allows for the precise determination of retention times and response factors in HPLC and LC-MS assays. Furthermore, knowing the exact conditions that favor this side reaction—such as the presence of free methyl mercaptan and specific Lewis acids—enables process engineers to modify the main API synthesis. They can implement scavengers or adjust stoichiometry to prevent the formation of the aluminum mercapto species in the main reactor, thereby reducing the burden of purification downstream and improving the overall yield of the desired Cefminox Sodium.
How to Synthesize Cefminox Impurity D Efficiently
The synthesis of Cefminox Impurity D requires precise control over reaction parameters to ensure the stability of the beta-lactam ring and the correct stereochemical outcome. The process is divided into five distinct stages, beginning with the preparation of the aluminum mercapto reagent and concluding with the final coupling and salt formation. Maintaining anhydrous conditions during the initial substitution and acylation steps is critical to prevent premature hydrolysis, while the final steps require careful pH adjustment to facilitate crystallization of the sodium salt. The detailed operational procedures, including specific molar ratios, solvent choices, and temperature profiles, are essential for reproducing the high purity reported in the patent examples. For laboratories aiming to produce this reference standard, adhering to the standardized protocol ensures consistency across batches.
- Generate aluminum mercapto by reacting aluminum trihalide with methyl mercaptan in a solvent like dichloromethane.
- React 7-MAC with the aluminum mercapto solution under organic phosphine catalysis to form the methoxy intermediate.
- Perform acylation with haloacetyl halide, followed by deprotection and final coupling with D-cysteine hydrochloride to yield the target impurity.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the ability to source or synthesize high-purity impurities like Cefminox Impurity D translates directly into risk mitigation and cost efficiency. The primary advantage of this patented route is the elimination of dependency on unpredictable isolation processes. Relying on extracting impurities from crude API batches creates a bottleneck; if the main production run has low levels of the impurity, the reference lab cannot generate enough standard for validation, stalling the entire quality control workflow. By adopting a dedicated synthetic route, companies secure a reliable supply of reference materials independent of API production volumes. This decoupling ensures that analytical development can proceed on schedule, regardless of the status of commercial manufacturing campaigns, thereby reducing lead time for high-purity reference standards and preventing costly delays in regulatory submissions.
- Cost Reduction in Manufacturing: While the synthesis of impurities incurs a cost, it is significantly lower than the hidden costs associated with failed regulatory audits or batch rejections due to uncharacterized peaks. By having a verified reference standard, quality control labs can accurately set specification limits, preventing the unnecessary rejection of safe API batches that merely contain acceptable levels of the impurity. Furthermore, the synthetic route uses readily available commodity chemicals like aluminum chloride and methyl mercaptan, avoiding the need for exotic catalysts or expensive enzymatic processes. The high yield and purity achieved in the described examples mean less solvent and energy are consumed per gram of purified product compared to preparative HPLC isolation, driving down the unit cost of the reference material substantially.
- Enhanced Supply Chain Reliability: The reliance on a defined chemical synthesis rather than biological fermentation byproducts or variable crude streams enhances supply chain resilience. The raw materials for this synthesis are stable, shelf-stable commodities with multiple global suppliers, reducing the risk of single-source bottlenecks. Additionally, the synthetic intermediate compounds generated along the route are more stable than the final beta-lactam impurity, allowing for the strategic stocking of precursors. This flexibility enables manufacturers to respond rapidly to surges in demand for reference standards, such as during a new drug application (NDA) filing or a regulatory inspection, ensuring that the supply chain remains robust and responsive to the dynamic needs of the pharmaceutical sector.
- Scalability and Environmental Compliance: The process described utilizes standard organic solvents like dichloromethane and methanol, which are easily recovered and recycled in modern chemical facilities, aligning with green chemistry principles. The reaction conditions are mild, operating near ambient temperatures, which reduces the energy footprint associated with heating or cryogenic cooling. Moreover, the high selectivity of the reaction minimizes the formation of complex waste streams, simplifying effluent treatment. From a scalability perspective, the chemistry is robust and does not rely on sensitive biocatalysts, making it straightforward to transfer from gram-scale laboratory synthesis to kilogram-scale production to support global regulatory needs without significant process re-engineering.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis and application of Cefminox Impurity D. These insights are derived directly from the technical specifications and experimental data provided in the patent literature, offering clarity on the feasibility and benefits of this synthetic approach for industry stakeholders.
Q: Why is synthesizing Cefminox Impurity D critical for regulatory approval?
A: Regulatory bodies require identified reference standards for all significant impurities found in the final API. Since Impurity D arises from specific side reactions during the methoxylation of 7-MAC, having a pure synthetic sample allows manufacturers to accurately quantify and control this impurity in their final Cefminox Sodium batches, ensuring patient safety and compliance.
Q: How does this patented route improve upon isolation from crude reaction mixtures?
A: Traditional isolation from crude API mixtures often yields impurities with purity levels below 80% due to the instability of beta-lactams and the complexity of the matrix. This dedicated synthetic route allows for the production of Impurity D with purity exceeding 97%, providing a reliable and stable reference material that does not degrade during storage or analysis.
Q: What are the key stability considerations for this compound?
A: As a beta-lactam derivative, Cefminox Impurity D is susceptible to hydrolysis and thermal degradation. The synthesis protocol emphasizes low-temperature conditions (often between -10°C and 20°C) and specific pH controls during the final coupling step to maintain the integrity of the bicyclic ring system and prevent decomposition.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Cefminox Impurity D Supplier
At NINGBO INNO PHARMCHEM, we understand that the integrity of your analytical data is only as good as the reference standards you use. Our team specializes in the custom synthesis of complex pharmaceutical intermediates and impurities, leveraging deep expertise in beta-lactam chemistry to deliver materials that meet the highest purity specifications. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that whether you need milligrams for method validation or grams for stability studies, we can deliver with precision. Our rigorous QC labs employ state-of-the-art NMR, MS, and HPLC technologies to certify every batch, guaranteeing that the structural identity and purity of our Cefminox Impurity D match the stringent requirements of global pharmacopoeias.
We invite you to collaborate with us to optimize your supply chain for critical reference materials. By partnering with our technical procurement team, you can request a Customized Cost-Saving Analysis to evaluate the economic benefits of switching from isolation to synthetic sourcing for your impurity standards. Contact us today to obtain specific COA data and route feasibility assessments tailored to your project timelines, and let us help you secure the quality and compliance of your Cefminox Sodium products.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →