Advanced Synthesis of Apremilast EP Impurity C for Global Pharmaceutical Quality Control
Introduction to Advanced Impurity Synthesis
The pharmaceutical industry faces increasing regulatory scrutiny regarding the purity and safety of active pharmaceutical ingredients (APIs), particularly for complex small molecules like Apremilast, a selective phosphodiesterase 4 (PDE4) inhibitor marketed as Otezla. Patent CN112876403A, published in June 2021, introduces a groundbreaking preparation method for Apremilast European Pharmacopoeia Impurity C, a critical reference standard required for rigorous quality control and impurity profiling. This patent addresses a significant gap in the prior art, where no published synthetic routes for this specific impurity existed, thereby hindering comprehensive quality research. By establishing a reliable pathway to generate this impurity, manufacturers can now accurately quantify related substances in bulk drugs, ensuring compliance with stringent global pharmacopoeial standards. The strategic importance of this technology cannot be overstated, as it empowers quality assurance teams to validate the safety profile of the final medication with unprecedented precision. 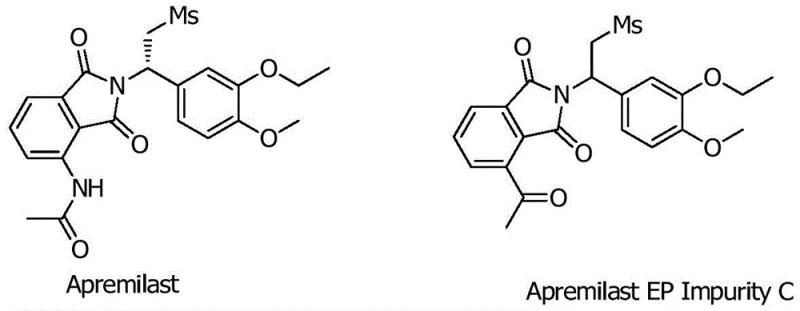
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior to the disclosure of this patent, the synthesis of Apremilast EP Impurity C was largely undocumented, forcing pharmaceutical companies to rely on inefficient isolation from crude reaction mixtures or non-existent commercial sources. Traditional approaches to synthesizing similar phthalimide derivatives often suffer from low regioselectivity, requiring extensive chromatographic purification that drastically reduces overall yield and increases production costs. Furthermore, existing methods for introducing the specific acetyl group at the ortho-position of the phthalimide ring frequently involve harsh conditions that compromise the integrity of sensitive functional groups, such as the methoxy substituents on the phenyl ring. These limitations create a bottleneck in the supply chain for high-purity reference standards, delaying drug development timelines and complicating regulatory filings. Without a dedicated synthetic route, the ability to perform accurate limit tests for this specific genotoxic or process-related impurity remains severely compromised, posing a risk to patient safety and market authorization.
The Novel Approach
The novel approach detailed in CN112876403A overcomes these historical challenges through a rational, convergent synthetic strategy that prioritizes modularity and high yield. The route begins with the oxidation of readily available 3-halogenated-2-methylbenzoic acid to a dicarboxylic acid, followed by dehydration to form a reactive anhydride, which serves as a robust building block for the phthalimide core. Simultaneously, a separate stream converts 3,4-dihydroxy benzonitrile into a functionalized amine via methylation and nucleophilic addition with dimethyl sulfone, ensuring precise control over the side-chain architecture. The convergence of these two intermediates through an amide exchange reaction efficiently constructs the core imide structure, setting the stage for the final transformation. 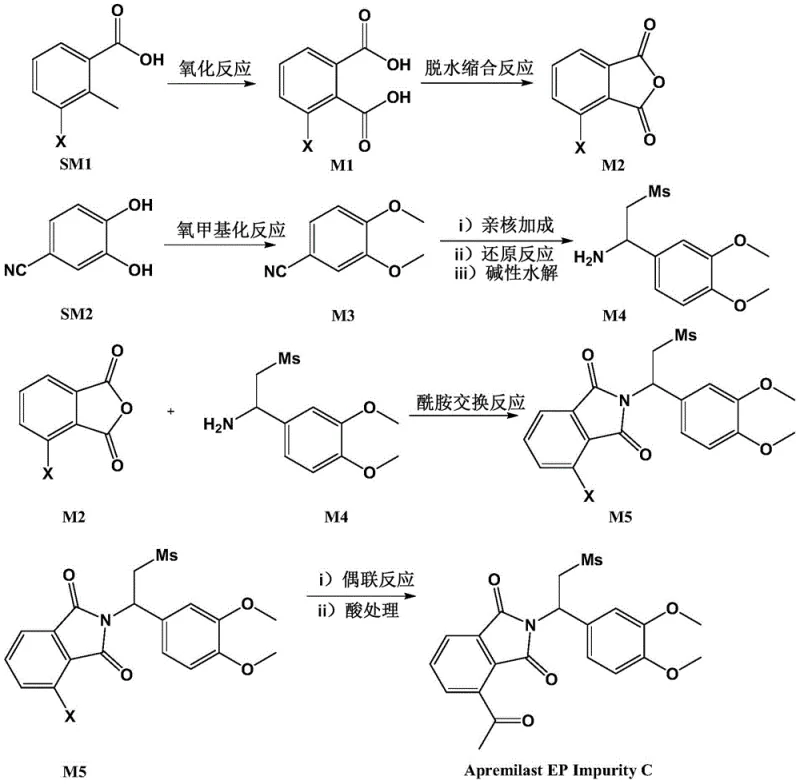
Mechanistic Insights into Pd-Catalyzed Carbonylation and Oxidation
The cornerstone of this synthetic innovation lies in the final step, which employs a sophisticated palladium-catalyzed coupling reaction to install the acetyl functionality with high fidelity. In this transformation, the bromo-substituted phthalimide intermediate reacts with tributyl(1-ethoxyvinyl)tin in the presence of a dichlorotriphenylphosphine palladium catalyst, facilitating a Stille-type coupling that introduces the vinyl ether moiety. Subsequent acid hydrolysis cleaves the enol ether to reveal the target methyl ketone, a reaction sequence that is remarkably tolerant of the surrounding electron-rich aromatic systems. This mechanistic pathway avoids the use of aggressive organolithium reagents at the final stage, which could otherwise attack the imide carbonyls, thereby preserving the structural integrity of the molecule. The careful selection of reaction conditions, including refluxing in toluene followed by mild acidic workup in methanol, demonstrates a deep understanding of chemoselectivity, ensuring that the final product is obtained with minimal byproduct formation. Such mechanistic precision is vital for producing reference standards that meet the exacting purity requirements of analytical laboratories worldwide.
Equally critical is the initial oxidation step, where 3-halogenated-2-methylbenzoic acid is converted to the corresponding phthalic acid derivative using potassium permanganate in an alkaline aqueous medium. This oxidation proceeds through a radical mechanism that selectively targets the benzylic methyl group while leaving the halogen substituent intact, a feat that requires precise control of temperature and pH to prevent over-oxidation or dehalogenation. The subsequent dehydration condensation using acetic anhydride cyclizes the dicarboxylic acid into the anhydride, activating the carboxyl groups for the subsequent nucleophilic attack by the amine intermediate. This sequence exemplifies classic organic synthesis principles applied with modern efficiency, utilizing inexpensive oxidants and dehydrating agents to drive the reaction to completion. The robustness of these early-stage transformations ensures that the bulk of the molecular complexity is established early in the process, minimizing the risk of failure in later, more valuable steps.
How to Synthesize Apremilast EP Impurity C Efficiently
The synthesis of this high-value impurity is designed for operational simplicity, utilizing common laboratory reagents and standard equipment to facilitate easy adoption by contract research organizations and internal QC departments. The process flows logically from simple aromatic acids and phenols, avoiding the need for exotic catalysts or cryogenic conditions beyond the initial lithiation step. Detailed standard operating procedures for each transformation, including specific molar ratios and temperature profiles, are provided in the patent to ensure reproducibility across different manufacturing sites. For a comprehensive, step-by-step guide on executing this synthesis with optimal yield and purity, please refer to the standardized protocol outlined below.
- Oxidize 3-halogenated-2-methylbenzoic acid to a dicarboxylic acid intermediate, followed by dehydration to form the anhydride.
- Methylate 3,4-dihydroxy benzonitrile, then perform nucleophilic addition with dimethyl sulfone and reduction to obtain the amine intermediate.
- Condense the anhydride and amine to form the imide, followed by Pd-catalyzed coupling and acid treatment to yield the final ketone impurity.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement perspective, the methodology described in this patent offers substantial advantages by leveraging commodity chemicals as starting materials, which significantly mitigates supply chain risks associated with specialized precursors. The reliance on 3-halogenated-2-methylbenzoic acid and 3,4-dihydroxy benzonitrile ensures that raw material availability is high and pricing is stable, shielding manufacturers from the volatility often seen in the fine chemical market. This stability is crucial for long-term supply agreements, as it guarantees consistent production capacity without the threat of raw material shortages disrupting the delivery of critical reference standards. Furthermore, the high yields reported in the patent examples suggest a highly efficient process that minimizes waste generation, aligning with modern green chemistry initiatives and reducing disposal costs. By adopting this route, supply chain managers can secure a reliable source of high-purity impurities that supports uninterrupted drug development and regulatory compliance activities.
- Cost Reduction in Manufacturing: The elimination of complex purification steps and the use of cost-effective reagents like potassium permanganate and acetic anhydride drive down the overall cost of goods sold. By avoiding expensive transition metal catalysts in the early stages and utilizing high-yielding condensation reactions, the process maximizes atom economy and reduces the financial burden of raw material consumption. This economic efficiency translates directly into lower prices for the end-user, making high-quality reference standards more accessible to generic drug manufacturers and research institutions. Additionally, the streamlined workflow reduces labor hours and equipment occupancy time, further enhancing the cost-effectiveness of the production campaign.
- Enhanced Supply Chain Reliability: The modular nature of the synthesis allows for the parallel production of key intermediates, which can be stockpiled to buffer against demand fluctuations or unexpected production delays. Since the starting materials are widely available from multiple global suppliers, the risk of single-source dependency is effectively eliminated, ensuring business continuity even in turbulent market conditions. This resilience is particularly valuable for pharmaceutical companies that require just-in-time delivery of impurity standards to support batch release testing and stability studies. The robustness of the chemical transformations also means that the process is less prone to batch failures, guaranteeing a steady flow of product to meet tight deadlines.
- Scalability and Environmental Compliance: The reaction conditions employed, such as aqueous oxidation and reflux in common organic solvents, are inherently scalable from gram to kilogram quantities without requiring specialized high-pressure or cryogenic infrastructure. This scalability facilitates the rapid transition from laboratory development to commercial manufacturing, allowing suppliers to respond quickly to surges in market demand. Moreover, the process generates manageable waste streams that can be treated using standard effluent handling protocols, ensuring compliance with increasingly strict environmental regulations. The absence of highly toxic reagents in the final steps further simplifies the safety profile of the manufacturing process, reducing the need for specialized containment facilities.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis and application of Apremilast EP Impurity C, based on the detailed disclosures within the patent literature. These insights are intended to clarify the practical implications of this new method for stakeholders involved in pharmaceutical quality control and process development. Understanding these nuances is essential for making informed decisions about sourcing strategies and analytical method validation.
Q: Why is Apremilast EP Impurity C critical for drug development?
A: Apremilast EP Impurity C is a specified impurity in the European Pharmacopoeia for Otezla. Controlling its levels is mandatory for regulatory approval and ensuring patient safety, making reliable reference standards essential for QC labs.
Q: What are the key advantages of the patented synthesis route?
A: The route described in CN112876403A utilizes readily available starting materials like 3-halogenated-2-methylbenzoic acid and avoids complex purification steps, resulting in high overall yield and purity suitable for commercial reference standard production.
Q: Can this synthesis be scaled for industrial production?
A: Yes, the process employs robust reactions such as oxidation with potassium permanganate and standard Pd-catalyzed coupling, which are well-established in industrial settings, facilitating easy scale-up from grams to kilograms.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Apremilast EP Impurity C Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-purity impurities play in the successful development and registration of generic and innovative drugs. Our team of expert chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet your volume requirements with consistent quality. We operate stringent purity specifications and maintain rigorous QC labs equipped with state-of-the-art analytical instrumentation to verify the identity and purity of every batch we produce. Our commitment to excellence means that when you partner with us, you are securing a supply of reference standards that will stand up to the most demanding regulatory audits.
We invite you to contact our technical procurement team to discuss your specific requirements for Apremilast EP Impurity C and other related reference standards. By engaging with us, you can request a Customized Cost-Saving Analysis tailored to your project's scale and timeline, helping you optimize your budget without compromising on quality. We encourage you to reach out today to obtain specific COA data and route feasibility assessments that will demonstrate our capability to support your drug development pipeline effectively.