Scalable Synthesis of 6-Fluoropenciclovir Intermediates for Antiviral API Production
Scalable Synthesis of 6-Fluoropenciclovir Intermediates for Antiviral API Production
The development of potent antiviral agents often hinges on the precise modification of nucleoside scaffolds, particularly through the introduction of halogen atoms to enhance biological activity and metabolic stability. Patent CN100569774C discloses a robust and industrially viable preparation method for 2-amino-6-fluoro-9-(4-hydroxy-3-hydroxymethylbutyl)purine, commonly known as 6-Fluoropenciclovir, and its key intermediates. This compound represents a critical evolution from the parent drug Penciclovir, offering improved binding affinity to viral thymidine kinase. The patented technology addresses significant bottlenecks in traditional synthesis routes, specifically focusing on the stabilization of reactive intermediates and the simplification of reagent handling. By replacing hazardous anhydrous gases with stable ethanolic solutions and optimizing thermal parameters, this methodology offers a pathway to high-purity pharmaceutical intermediates suitable for large-scale manufacturing.
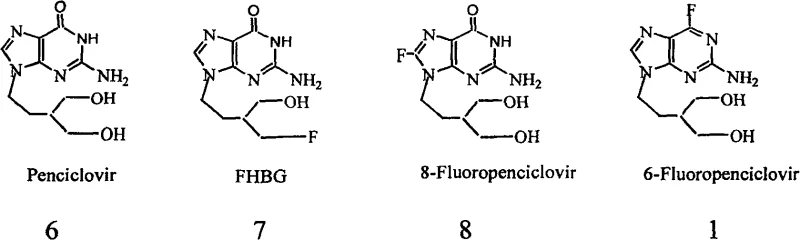
For procurement specialists and supply chain managers seeking a reliable pharmaceutical intermediate supplier, understanding the underlying chemical architecture is vital. The structural relationship between Penciclovir and its fluorinated analogs highlights the strategic placement of the fluorine atom at the C-6 position of the purine ring. This specific modification is not merely cosmetic; it fundamentally alters the electronic properties of the molecule, potentially enhancing its efficacy against herpes simplex viruses. The ability to synthesize this specific analog efficiently translates directly into cost reduction in API manufacturing, as it streamlines the production of next-generation antiviral therapeutics. The patent provides a comprehensive blueprint that moves away from laboratory-scale curiosities toward processes that can withstand the rigors of commercial production environments.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art methods, such as those described by Kim et al., relied heavily on the use of anhydrous trimethylamine gas for the quaternization step. This approach presents severe logistical and safety challenges for industrial application. Trimethylamine has an extremely low boiling point of approximately 2 to 3°C, necessitating complex cryogenic condensation systems to maintain the reagent in a liquid state during addition. Such conditions require specialized equipment capable of sustaining temperatures well below zero, significantly increasing capital expenditure and energy consumption. Furthermore, the reaction times associated with these legacy methods were excessively long, often extending up to five days to reach completion. This prolonged exposure increases the risk of side reactions and complicates batch scheduling, creating bottlenecks that hinder the commercial scale-up of complex pharmaceutical intermediates. Additionally, the direct sourcing of certain precursors used in these older routes was difficult, as they were not standard commercial reagents, further straining the supply chain.
The Novel Approach
The innovative process outlined in the patent fundamentally re-engineers the synthesis to overcome these thermal and logistical hurdles. A pivotal improvement is the substitution of anhydrous trimethylamine gas with a commercially available trimethylamine ethanol solution. This simple change eliminates the need for cryogenic condensation, allowing the reaction to proceed under much milder conditions, typically initiating between -20°C and 0°C before warming to room temperature. This shift drastically reduces the reaction time from several days to merely overnight, representing a massive gain in throughput efficiency. The stability of the resulting nucleoside ammonium salt intermediate is carefully managed by controlling the fluorination temperature between 20°C and 60°C. This precision prevents the thermal decomposition observed in previous methods, ensuring that the reaction yields the desired 6-fluoro product with minimal by-product generation. The entire sequence, from the diacetyl precursor to the final fluorinated purine, is designed for operational simplicity and high reproducibility.
Mechanistic Insights into Quaternization and Nucleophilic Fluorination
The core of this synthetic strategy lies in the activation of the purine ring through quaternization, which facilitates the subsequent nucleophilic aromatic substitution. In the conversion of Compound 3 to Compound 4, trimethylamine acts as a nucleophile, attacking the electron-deficient purine ring to form a quaternary ammonium salt. This positive charge on the ring nitrogen significantly enhances the electrophilicity of the C-6 carbon, making the chlorine atom at this position much more susceptible to displacement. Unlike traditional methods where the leaving group ability of chlorine might be insufficient under mild conditions, the quaternary intermediate lowers the activation energy for the substitution reaction. This mechanistic nuance allows the use of potassium fluoride, a relatively mild and inexpensive fluorinating agent, instead of more aggressive and hazardous reagents. The stability of this ammonium salt is temperature-dependent; maintaining the environment below 60°C ensures the integrity of the activated complex, whereas higher temperatures lead to degradation pathways that compromise the purity of the final API intermediate.
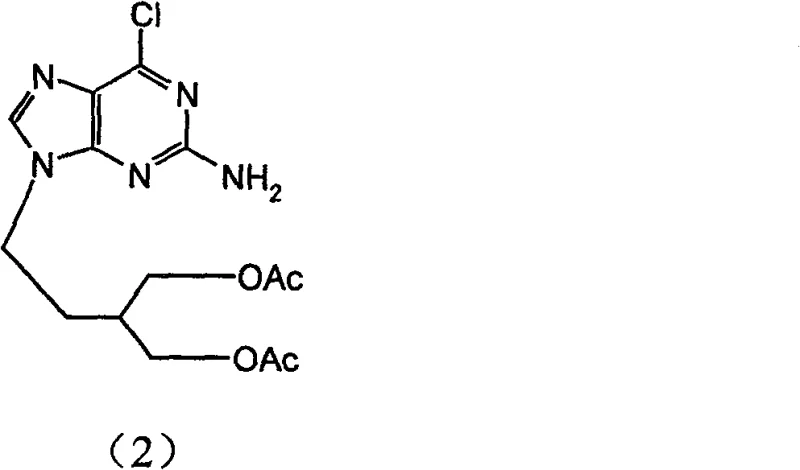
Impurity control is rigorously addressed through the optimization of the hydrolysis and fluorination steps. During the initial hydrolysis of the diacetyl precursor (Compound 2), the use of potassium carbonate in a methanol-water mixture at controlled temperatures (0-20°C) ensures selective deprotection of the acetyl groups without affecting the 6-chloro substituent. This chemoselectivity is crucial for preventing the formation of des-chloro impurities early in the synthesis. In the final fluorination step, the use of anhydrous potassium fluoride in dimethylformamide (DMF) provides a source of naked fluoride ions that efficiently displace the activated chlorine. The patent data indicates that deviations in temperature, specifically exceeding 60°C, trigger decomposition of the intermediate salt, leading to complex mixtures that are difficult to purify. By adhering to the specified thermal window, manufacturers can achieve high-purity 6-Fluoropenciclovir with yields exceeding 87%, demonstrating the robustness of the mechanistic design against process variations.
How to Synthesize 6-Fluoropenciclovir Efficiently
The synthesis of this high-value antiviral intermediate requires strict adherence to the optimized protocol to ensure maximum yield and purity. The process begins with the careful hydrolysis of the protected precursor, followed by the critical quaternization step that activates the molecule for fluorination. Each stage demands precise control over solvent ratios, temperature gradients, and reaction monitoring via thin-layer chromatography (TLC). The following guide outlines the standardized operational procedure derived from the patent examples, providing a clear roadmap for technical teams aiming to implement this route. For detailed execution parameters and specific stoichiometric ratios, please refer to the structured synthesis guide below.
- Hydrolyze the diacetyl precursor (Compound 2) using potassium carbonate in a methanol-water mixture at 0-20°C to obtain the diol intermediate (Compound 3).
- React Compound 3 with trimethylamine ethanol solution in a THF/DMF solvent system, starting at -20°C and warming to room temperature to form the stable nucleoside ammonium salt (Compound 4).
- Perform nucleophilic fluorination on Compound 4 using anhydrous potassium fluoride in DMF at 60°C to yield the final 6-Fluoropenciclovir (Compound 1).
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, this patented methodology offers substantial advantages that directly impact the bottom line and supply chain resilience. The transition from gas-phase reagents to liquid solutions simplifies the procurement landscape, as trimethylamine ethanol solution is a standard commodity chemical with a stable supply chain. This shift removes the dependency on specialized gas handling infrastructure, thereby reducing both capital investment and ongoing maintenance costs for production facilities. Furthermore, the reduction in reaction time from days to hours significantly increases asset utilization, allowing manufacturers to produce more batches within the same timeframe without expanding their physical footprint. These operational efficiencies translate into tangible economic benefits, making the production of this specialized nucleoside analog more financially viable for generic and branded drug manufacturers alike.
- Cost Reduction in Manufacturing: The elimination of cryogenic cooling systems represents a major saving in energy consumption and equipment costs. Handling anhydrous gases requires expensive condensers and safety protocols that are not needed when using ethanolic solutions. Additionally, the use of common inorganic salts like potassium carbonate and potassium fluoride keeps raw material costs low compared to exotic organometallic reagents. The high yields reported in the patent examples suggest minimal waste generation, further optimizing the cost per kilogram of the final product. These factors combined create a highly competitive cost structure for the production of high-purity pharmaceutical intermediates.
- Enhanced Supply Chain Reliability: By utilizing readily available solvents such as tetrahydrofuran (THF), dimethylformamide (DMF), and ethanol, the process mitigates the risk of raw material shortages. The reagents involved are produced at a global scale, ensuring consistent availability even during market fluctuations. The robustness of the reaction conditions, which do not require extreme pressures or temperatures, means that production can be easily transferred between different manufacturing sites without significant requalification efforts. This flexibility is crucial for maintaining reducing lead time for high-purity pharmaceutical intermediates and ensuring uninterrupted supply to downstream API producers.
- Scalability and Environmental Compliance: The process is inherently scalable, moving seamlessly from gram-scale laboratory experiments to multi-ton commercial production. The absence of high-pressure steps and the use of standard filtration and column chromatography for purification simplify the engineering requirements for scale-up. Moreover, the simplified workup procedures reduce the volume of solvent waste generated per unit of product. The ability to operate at near-ambient pressures and moderate temperatures aligns well with modern green chemistry principles, facilitating easier compliance with environmental regulations and reducing the burden on waste treatment facilities.
Frequently Asked Questions (FAQ)
The following questions address common technical and operational inquiries regarding the synthesis of 6-Fluoropenciclovir. These answers are derived directly from the experimental data and technical specifications provided in the patent documentation. They are intended to clarify the critical process parameters that ensure successful implementation of this technology. Understanding these nuances is essential for process engineers and quality assurance teams tasked with validating the manufacturing route.
Q: Why is the trimethylamine ethanol solution preferred over anhydrous gas?
A: Using trimethylamine ethanol solution eliminates the need for complex cryogenic condensation equipment required for handling anhydrous trimethylamine gas (boiling point 2-3°C). This significantly simplifies the operational setup and reduces safety risks associated with high-pressure gas cylinders.
Q: What is the critical temperature control parameter for the fluorination step?
A: The reaction temperature must be maintained between 20°C and 60°C. Temperatures exceeding 60°C cause the unstable nucleoside ammonium salt intermediate to decompose, leading to significant by-product formation and reduced yield.
Q: How does this method improve impurity profiles compared to prior art?
A: By stabilizing the intermediate ammonium salt and avoiding harsh thermal conditions, the process minimizes side reactions. The use of mild hydrolysis conditions also preserves the 6-chloro functionality until the specific fluorination step, ensuring higher regioselectivity.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 6-Fluoropenciclovir Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the development of life-saving antiviral medications. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from pilot plant to full-scale manufacturing is seamless and efficient. We are committed to delivering products that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation. Our expertise in nucleoside chemistry allows us to navigate the complexities of fluorination and quaternization reactions with precision, guaranteeing a consistent supply of material that meets the exacting standards of the global pharmaceutical industry.
We invite potential partners to engage with our technical procurement team to discuss how this optimized synthesis route can benefit your specific project requirements. By leveraging our manufacturing capabilities, you can achieve significant process improvements and secure a stable supply of critical materials. We encourage you to request a Customized Cost-Saving Analysis tailored to your volume needs. Our team is ready to provide specific COA data and route feasibility assessments to support your regulatory filings and production planning. Contact us today to explore how we can collaborate to bring advanced antiviral therapies to market faster and more efficiently.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →