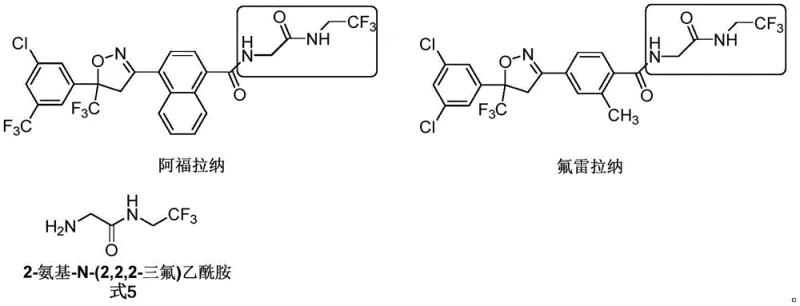
Advanced Synthesis of Fluralaner Key Fragments and Impurity Standards for Veterinary Pharma
The veterinary pharmaceutical landscape is undergoing a significant transformation, driven by the increasing demand for effective parasiticides to protect companion animals. Central to this evolution are isoxazoline-class drugs like Fluralaner and Affoxolaner, which have revolutionized the treatment of flea and tick infestations. However, the commercial success of these Active Pharmaceutical Ingredients (APIs) relies heavily on the purity and quality of their synthetic intermediates. Patent CN113121383A, published in July 2021, addresses a critical gap in this supply chain by disclosing a novel method for synthesizing and identifying related substances of a key building block: 2-amino-N-(2,2,2-trifluoroethyl) acetamide (Formula 5). This patent does not merely describe the synthesis of the intermediate itself but focuses on the identification and preparation of a specific, previously unknown impurity, 2-amino-N,N',N'-triglycyl acethydrazide (Formula 7). For R&D directors and quality assurance teams, this development is pivotal. It transforms an unidentified peak in chromatographic analysis into a characterized reference standard, thereby enabling rigorous quality control (QC) protocols that are essential for regulatory approval and market access.
The ability to synthesize this specific impurity standard allows manufacturers to validate their HPLC methods with unprecedented accuracy. In the context of global regulatory bodies like the FDA or EMA, the presence of unknown impurities above certain thresholds can halt the approval of a new drug application. By providing a synthetic route to Formula 7, this patent empowers manufacturers to demonstrate control over their process impurities, ensuring that the final veterinary drug products meet the highest safety standards for animal health.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditionally, the synthesis of complex fluorinated intermediates like 2-amino-N-(2,2,2-trifluoroethyl) acetamide has been plagued by challenges in impurity profiling. During the scale-up of such intermediates, side reactions often occur, leading to the formation of trace byproducts that are difficult to identify. In many conventional workflows, these byproducts appear as "unknown peaks" in High-Performance Liquid Chromatography (HPLC) spectra. Without a reference standard, it is impossible to determine the exact chemical structure of these impurities, let alone quantify them accurately. This uncertainty creates a significant bottleneck in the drug development lifecycle. Quality control teams are forced to rely on relative retention times or non-specific detection methods, which may not satisfy the rigorous demands of modern pharmacopoeias. Furthermore, the inability to pinpoint the source of an impurity makes process optimization difficult; engineers cannot effectively tweak reaction parameters to suppress a byproduct if they do not understand its formation mechanism. This lack of clarity often leads to batch-to-batch variability, increased waste, and potential delays in regulatory filings due to insufficient impurity characterization data.
The Novel Approach
The approach detailed in patent CN113121383A represents a paradigm shift from reactive problem-solving to proactive quality design. Instead of ignoring unknown peaks, the inventors have successfully isolated and synthesized the specific impurity, Formula 7, allowing it to be used as a definitive reference standard. This strategy turns a potential liability into a tool for precision. By having the authentic substance of the impurity available, analytical chemists can spike their samples to confirm peak identity, calibrate their detectors for accurate quantification, and set scientifically justified acceptance criteria. The patent outlines a robust two-step synthesis starting from 2-(1,3-dioxoisoindol-2-yl) acetic acid, utilizing acetic anhydride and hydrazine hydrate. This route is distinct from the main API synthesis, designed specifically to generate the impurity structure efficiently. This capability ensures that manufacturers of Fluralaner and Affoxolaner intermediates can implement a "quality by design" (QbD) framework, where every component of the chemical profile is understood and controlled. Consequently, this leads to more stable manufacturing processes and a smoother path through regulatory audits.
Mechanistic Insights into Impurity Formation and Control
To fully appreciate the value of this patent, one must understand the chemical logic behind the synthesis of both the target intermediate and its associated impurity. The core intermediate, 2-amino-N-(2,2,2-trifluoroethyl) acetamide (Formula 5), is typically synthesized via a Gabriel synthesis-like pathway. As illustrated in the reaction scheme below, the process begins with the condensation of phthalic anhydride and glycine to form 2-(1,3-dioxoisoindol-2-yl) acetic acid (Formula 3). This protected amino acid is then activated and coupled with a trifluoroethyl amine derivative to form the protected amide (Formula 4). Finally, hydrazine hydrate is employed to cleave the phthalimide protecting group, releasing the free amine (Formula 5). This multi-step sequence requires precise control over stoichiometry and reaction conditions to prevent side reactions.
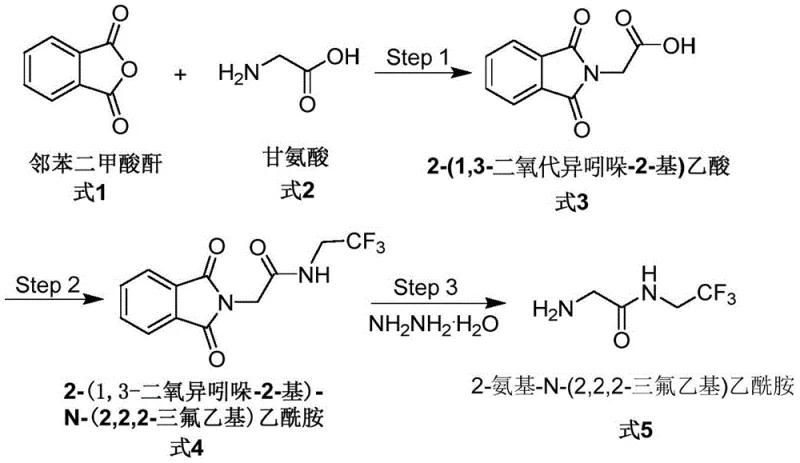
The formation of the impurity, 2-amino-N,N',N'-triglycyl acethydrazide (Formula 7), is a fascinating example of how reagents intended for deprotection can participate in alternative condensation pathways. The patent reveals that this impurity arises from the interaction of glycine-derived fragments with hydrazine. Specifically, the invention demonstrates that by reacting the anhydride derivative of the protected glycine (Formula 6) directly with excess hydrazine hydrate, one can construct the tetra-substituted hydrazide core of Formula 7. This mechanistic insight is crucial for R&D teams. It suggests that during the final deprotection step of the main synthesis (Step 3 in the main route), if the concentration of hydrazine is too high or if local hotspots of the activated acid species exist, there is a risk of oligomerization or condensation leading to Formula 7. Understanding this mechanism allows process chemists to mitigate the risk by controlling the addition rate of hydrazine, optimizing the temperature profile, or adjusting the solvent system to minimize the formation of this specific byproduct. The ability to synthesize Formula 7 independently confirms its structure via NMR and single-crystal diffraction, removing any ambiguity about its identity in the final product mixture.
How to Synthesize 2-amino-N,N',N'-triglycyl acethydrazide Efficiently
The synthesis of the impurity standard described in this patent offers a reliable protocol for generating high-purity reference materials. The process leverages readily available starting materials and standard unit operations, making it accessible for most fine chemical laboratories. The initial step involves the activation of the carboxylic acid group using acetic anhydride, creating a highly reactive mixed anhydride species. This intermediate is then subjected to nucleophilic attack by hydrazine, which acts as both a deprotecting agent and a coupling partner to form the central hydrazide linkage. The subsequent workup involves careful pH control and crystallization to ensure the removal of unreacted starting materials and byproducts.
- Condense 2-(1,3-dioxoisoindol-2-yl) acetic acid with acetic anhydride at 100°C for 24 hours to form the reactive anhydride intermediate.
- React the resulting anhydride with hydrazine hydrate in ethanol under reflux at 80°C for 10 hours to generate the crude triglycyl acethydrazide.
- Purify the final product through filtration, solvent removal, and pH adjustment to isolate the white solid impurity standard.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the implications of this patent extend far beyond the laboratory bench. The availability of a validated synthesis for impurity standards translates directly into supply chain resilience and cost efficiency. When a manufacturer can internally produce their own QC standards, they reduce their dependency on external specialty chemical suppliers who often charge exorbitant prices for small quantities of reference materials. This vertical integration capability significantly lowers the operational costs associated with quality assurance. Moreover, having immediate access to these standards accelerates the release testing of batches. Instead of waiting weeks for a third-party vendor to ship a reference standard, QC labs can verify their results in real-time, reducing the lead time for product release and improving inventory turnover rates.
- Cost Reduction in Manufacturing: The ability to synthesize impurity standards in-house eliminates the need for expensive external sourcing of rare reference compounds. This internal capability prevents production delays caused by the unavailability of critical QC materials. Furthermore, by understanding the mechanism of impurity formation, manufacturers can optimize their main synthesis process to minimize waste and improve overall yield. Reducing the formation of byproducts like Formula 7 means less raw material is lost to side reactions, directly improving the cost-of-goods-sold (COGS) for the key intermediate. The process described uses common reagents like acetic anhydride and hydrazine hydrate, which are commodity chemicals with stable pricing, further insulating the supply chain from volatile market fluctuations associated with exotic reagents.
- Enhanced Supply Chain Reliability: Regulatory compliance is the backbone of a reliable supply chain in the pharmaceutical industry. By adopting the methods disclosed in CN113121383A, suppliers can provide comprehensive impurity profiles with their Certificates of Analysis (COA). This level of transparency builds trust with downstream API manufacturers and ultimately with the veterinary drug companies. It mitigates the risk of batch rejection at the customer's site, which can be catastrophic for supply continuity. A supplier who can guarantee the absence of unknown impurities—or precisely quantify them against a known standard—becomes a preferred partner. This reliability reduces the need for safety stock and allows for leaner inventory management strategies across the entire value chain, from intermediate synthesis to final drug formulation.
- Scalability and Environmental Compliance: The synthesis route for the impurity standard is designed with scalability in mind. It avoids the use of hazardous transition metal catalysts or extreme pressure conditions, relying instead on thermal activation in common solvents like ethanol and acetic anhydride. This simplicity facilitates easy scale-up from gram-scale laboratory synthesis to kilogram-scale production without requiring specialized equipment. From an environmental perspective, the process generates manageable waste streams. The byproducts are primarily organic acids and salts that can be treated using standard wastewater protocols. This alignment with green chemistry principles ensures that the manufacturing process remains compliant with increasingly stringent environmental regulations, avoiding potential fines or shutdowns that could disrupt supply.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the synthesis and application of these veterinary drug intermediates. The answers are derived directly from the experimental data and structural analysis provided in the patent documentation. Understanding these details is essential for technical teams evaluating the feasibility of integrating this intermediate into their production workflows.
Q: What is the primary application of 2-amino-N-(2,2,2-trifluoroethyl) acetamide?
A: This compound serves as a critical building block (Formula 5) in the synthesis of isoxazoline insecticides and acaricides such as Fluralaner and Affoxolaner, which are widely used in veterinary medicine for treating parasitic infections in dogs.
Q: Why is the synthesis of Impurity Formula 7 significant for quality control?
A: Identifying and synthesizing the specific impurity 2-amino-N,N',N'-triglycyl acethydrazide (Formula 7) allows manufacturers to establish precise HPLC reference standards. This ensures that trace impurities in the final API can be accurately quantified and controlled, meeting stringent regulatory requirements.
Q: What are the reaction conditions for synthesizing the impurity standard?
A: The synthesis involves heating 2-(1,3-dioxoisoindol-2-yl) acetic acid in acetic anhydride at 100°C, followed by a reaction with hydrazine hydrate in ethanol at reflux (80°C). This robust protocol yields a high-purity reference material suitable for analytical calibration.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-amino-N-(2,2,2-trifluoroethyl) acetamide Supplier
The technological advancements detailed in patent CN113121383A highlight the complexity and precision required in modern veterinary drug synthesis. At NINGBO INNO PHARMCHEM, we recognize that delivering high-quality intermediates is about more than just chemical transformation; it is about providing total quality assurance. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet the volumetric demands of global pharmaceutical partners. We operate with stringent purity specifications and utilize rigorous QC labs equipped to detect and quantify trace impurities down to the ppm level. Our commitment to "Quality by Design" means we don't just sell chemicals; we deliver validated solutions that streamline your regulatory filing process.
We invite procurement leaders and R&D directors to collaborate with us on optimizing their supply chains for Fluralaner and Affoxolaner precursors. By leveraging our technical expertise, you can secure a stable supply of high-purity intermediates that meet the most demanding international standards. Contact our technical procurement team today to request a Customized Cost-Saving Analysis. We are ready to provide specific COA data and route feasibility assessments tailored to your project's unique requirements, ensuring your path to market is efficient and compliant.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →