Scalable Synthesis of Nifuratel Related Substance A Hydrochloride for Advanced Impurity Profiling
The pharmaceutical industry faces relentless pressure to ensure the highest standards of drug safety, particularly concerning the identification and quantification of impurities in active pharmaceutical ingredients (APIs). In the context of Nifuratel, a broad-spectrum antibiotic widely used for gynecological infections, the control of related substances is paramount for regulatory compliance and patient safety. Patent CN112745271A introduces a groundbreaking preparation method for Nifuratel Related Substance A hydrochloride, addressing the critical shortage of high-purity reference standards. This innovation shifts the paradigm from difficult isolation techniques to a robust, four-step synthetic route starting from epichlorohydrin. By establishing a reliable supply of this specific impurity standard, manufacturers can now implement more rigorous quality control protocols, ensuring that final drug products meet the stringent purity requirements set by global health authorities. The technical significance of this development extends beyond mere compliance; it empowers R&D teams to better understand the degradation pathways of Nifuratel, ultimately leading to more stable formulations and improved therapeutic outcomes for patients worldwide.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the acquisition of Nifuratel Related Substance A hydrochloride has been a significant bottleneck in the quality assurance workflows of pharmaceutical manufacturers. The conventional approach primarily relied on the preparative liquid phase isolation of this substance directly from Nifuratel reaction liquids. This method is inherently flawed due to the complex matrix of the reaction mixture, which contains numerous by-products and unreacted starting materials. Consequently, the isolation process is characterized by extremely low yields, making it economically unviable for large-scale reference standard production. Furthermore, the separation difficulty is high, often requiring multiple rounds of chromatography which increases both the cost and the time required to obtain the substance. The resulting material often suffers from inconsistent purity levels, which compromises its utility as a quantitative standard in HPLC analysis. For supply chain managers, this reliance on isolation creates a fragile dependency on the production batches of the API itself, leading to unpredictable availability and potential delays in critical impurity profiling studies.
The Novel Approach
In stark contrast to the archaic isolation techniques, the novel synthetic route disclosed in the patent offers a streamlined and efficient pathway to the target molecule. This method decouples the production of the impurity standard from the API manufacturing process, allowing for dedicated, optimized synthesis. The route begins with the etherification of epichlorohydrin, a commodity chemical, followed by hydrazinolysis and a subsequent cyclization with diethyl carbonate. This logical progression builds the oxazolidinone core with high precision. The most striking advantage is the dramatic improvement in yield and purity; experimental data indicates purities reaching up to 99.89% with yields significantly surpassing those of isolation methods. For procurement specialists, this translates to a more stable cost structure and guaranteed availability. The process eliminates the need for complex preparative HPLC separations, replacing them with standard crystallization and filtration unit operations that are easily scalable. This shift not only enhances the reliability of the supply chain for high-purity pharmaceutical intermediates but also aligns perfectly with the industry's move towards greener, more atom-economical manufacturing processes.
Mechanistic Insights into the Four-Step Synthetic Route
The core of this technological breakthrough lies in the precise control of reaction conditions across four distinct chemical transformations. The first step involves the nucleophilic substitution of epichlorohydrin with sodium methoxide. Crucially, the addition of 18-crown-6 as a phase transfer catalyst (0.05 to 0.2 molar equivalents) facilitates the solubility of the methoxide ion in the organic phase, driving the reaction to completion at mild temperatures of 0-10°C. This prevents the polymerization of the epoxide ring, a common side reaction. The second step utilizes hydrazine hydrate to open the oxetane ring formed in the previous step, generating the hydrazine intermediate. This reaction is conducted at elevated temperatures (80-100°C) to overcome the activation energy barrier, ensuring complete conversion. The third step is the cyclization reaction where diethyl carbonate acts as the carbonyl source. Under the influence of an alkaline catalyst like sodium ethoxide, the hydrazine nitrogen attacks the carbonate carbonyl, closing the ring to form the 2-oxazolidinone structure. Finally, the free base is converted to the hydrochloride salt through controlled acidification, which enhances the stability and crystallinity of the final product.
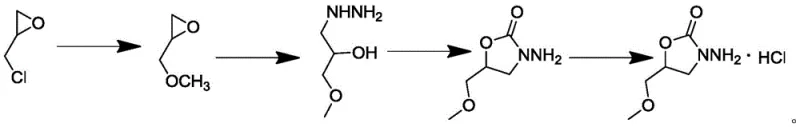
Impurity control within this synthetic sequence is achieved through careful optimization of stoichiometry and temperature profiles. For instance, maintaining the initial etherification at low temperatures minimizes the formation of polymeric by-products. In the cyclization step, the use of specific molar equivalents of diethyl carbonate (1.0 to 1.2 equivalents) ensures that the reaction proceeds without excess reagent that could lead to urea formation or other carbonate-derived impurities. The purification strategy relies heavily on the differential solubility of the product in various solvents. The patent highlights the use of dispersing solvents like dichloromethane or ethyl acetate, followed by salification with concentrated hydrochloric acid. This induces crystallization of the hydrochloride salt, leaving soluble impurities in the mother liquor. A final pulping step with absolute ethanol further washes away surface impurities, resulting in the reported high purity levels. This mechanistic understanding allows process chemists to troubleshoot effectively and maintain consistent quality across different production batches, a critical factor for R&D directors overseeing analytical method validation.
How to Synthesize Nifuratel Related Substance A Hydrochloride Efficiently
Implementing this synthesis requires adherence to specific operational parameters to maximize yield and safety. The process is designed to be compatible with standard glass-lined or stainless steel reactors found in most fine chemical facilities. Operators must pay close attention to the exothermic nature of the initial etherification and the hydrazinolysis steps, utilizing appropriate cooling systems to maintain the specified temperature ranges. The use of 18-crown-6, while effective, requires careful handling and recovery protocols to minimize costs. The subsequent steps involve standard work-up procedures such as phase separation, evaporation, and crystallization, which are well-understood unit operations in the industry. For detailed procedural specifics regarding reagent addition rates, stirring speeds, and drying parameters, operators should refer to the standardized technical documentation provided below.
- React sodium methoxide with epichlorohydrin using 18-crown-6 catalyst at 0-10°C to form 2-(methoxymethyl)-oxetane.
- Perform hydrazinolysis by adding the oxetane intermediate into hydrazine hydrate at 80-100°C to yield 3-methoxy-2-hydroxy-propylhydrazine.
- Cyclize the hydrazine derivative with diethyl carbonate and an alkaline catalyst at 50-85°C to obtain N-amino-5-methoxymethyl-2-oxazolidinone.
- Form the hydrochloride salt by treating the oxazolidinone with concentrated hydrochloric acid in a dispersing solvent, followed by crystallization and drying.
Commercial Advantages for Procurement and Supply Chain Teams
The transition from isolation to direct synthesis offers profound commercial benefits that extend well beyond the laboratory bench. For procurement managers, the ability to source this critical reference standard from a dedicated synthetic route mitigates the risk of supply disruption associated with API production schedules. The use of commodity starting materials like epichlorohydrin and diethyl carbonate ensures that raw material costs remain stable and predictable, shielding the supply chain from the volatility often seen with specialized precursors. Furthermore, the high yield of the process means that less raw material is wasted, contributing to a more sustainable and cost-effective operation. This efficiency allows suppliers to offer competitive pricing without compromising on the rigorous quality standards required for pharmaceutical applications. The robustness of the synthesis also implies a longer shelf-life for the production campaigns, enabling manufacturers to build strategic inventory buffers against market fluctuations.
- Cost Reduction in Manufacturing: The elimination of preparative liquid chromatography represents a massive reduction in operational expenditure. Traditional isolation methods consume vast amounts of expensive solvents and stationary phases, driving up the cost per gram exponentially. By replacing this with crystallization-based purification, the new method drastically lowers solvent consumption and waste disposal costs. Additionally, the high atom economy of the cyclization step ensures that the majority of the input mass is converted into the desired product, further optimizing the cost structure. This economic efficiency makes it feasible to produce the substance in multi-kilogram quantities, satisfying the demands of large-scale QC laboratories without the prohibitive costs previously associated with impurity standards.
- Enhanced Supply Chain Reliability: Dependence on API reaction liquors for impurity isolation creates a single point of failure in the supply chain. If API production is halted or delayed, the supply of the reference standard is immediately jeopardized. The independent synthetic route breaks this dependency, allowing for continuous production regardless of the API manufacturing status. The use of stable, commercially available reagents ensures that sourcing is straightforward and reliable. This decoupling enhances the overall resilience of the supply chain, ensuring that quality control teams always have access to the necessary standards to release batches of Nifuratel for the market, thereby preventing costly delays in product distribution.
- Scalability and Environmental Compliance: The reaction conditions employed in this synthesis are mild and easily scalable from gram to tonne scale. The temperatures range from 0°C to 100°C, which can be managed with standard industrial heating and cooling infrastructure. There is no requirement for high-pressure equipment or exotic catalysts that would complicate scale-up. From an environmental perspective, the reduction in solvent usage and the avoidance of complex chromatographic waste streams significantly lower the environmental footprint of the process. This aligns with modern green chemistry principles and simplifies regulatory compliance regarding waste management, making it an attractive option for manufacturers looking to improve their sustainability metrics while maintaining high production volumes.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of Nifuratel Related Substance A Hydrochloride. These insights are derived directly from the technical specifications and experimental data provided in the patent literature. Understanding these details is crucial for stakeholders involved in the procurement, quality assurance, and process development of Nifuratel and its related formulations. The answers reflect the current state-of-the-art in synthetic methodology for this specific pharmaceutical intermediate.
Q: Why is the synthesis of Nifuratel Related Substance A Hydrochloride critical for pharmaceutical quality control?
A: Nifuratel is known to be chemically unstable and can generate harmful impurities during synthesis and storage. Regulatory agencies require strict limits (typically <0.10%) for unknown impurities. Having a reliable synthetic source of Related Substance A allows manufacturers to accurately quantify and control this specific impurity, ensuring the safety and efficacy of the final drug product.
Q: What are the primary advantages of this new synthetic route compared to traditional isolation methods?
A: Traditional methods rely on isolating the substance from Nifuratel reaction liquids, which results in low yields and difficult separation processes. The novel four-step synthesis described in patent CN112745271A starts from readily available epichlorohydrin, offering significantly higher yields, superior purity (up to 99.89%), and a much more straightforward purification process suitable for industrial scaling.
Q: What are the key reaction conditions for the cyclization step in this process?
A: The critical cyclization step involves reacting 3-methoxy-2-hydroxy-propylhydrazine with diethyl carbonate. The process utilizes an alkaline catalyst, such as sodium ethoxide (0.1-0.4 molar equivalents), and maintains a reaction temperature between 50-85°C. These mild conditions facilitate the formation of the oxazolidinone ring while minimizing side reactions.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Nifuratel Related Substance A Hydrochloride Supplier
At NINGBO INNO PHARMCHEM, we recognize that the integrity of your pharmaceutical products depends on the quality of every component, including the reference standards used for testing. Our team of expert process chemists has thoroughly analyzed the synthetic route described in CN112745271A and is fully prepared to execute this technology at a commercial scale. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with consistency and precision. Our state-of-the-art facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, guaranteeing that every batch of Nifuratel Related Substance A Hydrochloride meets the highest industry standards. We are committed to being a partner in your quality assurance journey, providing the reliable materials necessary to safeguard public health.
We invite you to collaborate with us to optimize your supply chain for this critical intermediate. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements. Whether you need small quantities for method development or bulk supplies for routine QC, we encourage you to contact us to request specific COA data and route feasibility assessments. Let us help you secure a stable, high-quality supply of Nifuratel Related Substance A Hydrochloride, ensuring your operations run smoothly and efficiently.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →