Advanced Synthesis of Water-Soluble Rebeccamycin Analogues for Oncology Drug Development
Introduction to Novel Anticancer Intermediates
The landscape of oncology drug discovery is constantly evolving, driven by the need for compounds that combine potent biological activity with favorable physicochemical properties. Patent CN102898489B introduces a significant breakthrough in this domain by detailing the synthesis of novel N-6 substituted Rebeccamycin analogues. Rebeccamycin, originally isolated from Saccharothrix Aerocoligenes, is a known topoisomerase I inhibitor with strong antitumor potential. However, its clinical application has been historically hindered by poor water solubility. This patent addresses this critical bottleneck by modifying the indolocarbazole nucleus at the N-6 position with various hydrophilic substituents, including sugars, amino acids, and nucleic acid bases. 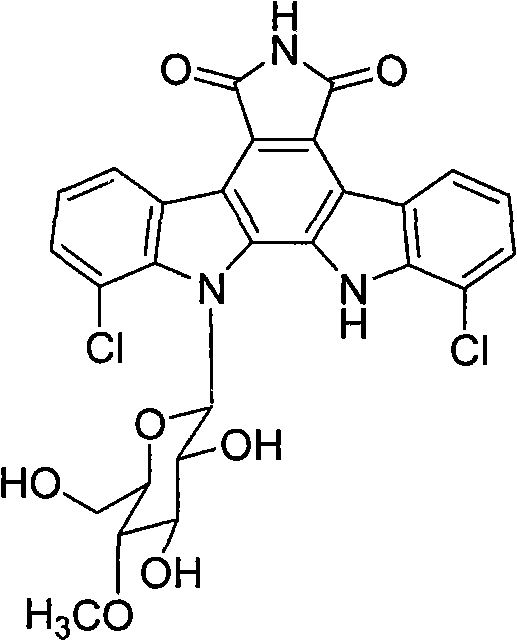 This strategic modification not only enhances solubility but also aims to improve targeting specificity and overall biological efficacy, presenting a valuable opportunity for pharmaceutical developers seeking next-generation anticancer agents.
This strategic modification not only enhances solubility but also aims to improve targeting specificity and overall biological efficacy, presenting a valuable opportunity for pharmaceutical developers seeking next-generation anticancer agents.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional approaches to utilizing Rebeccamycin have faced substantial hurdles primarily due to the molecule's inherent hydrophobicity. The natural compound consists of an indole[2,3-a]pyrrole[3,4-c]carbazole nucleus and a beta-glucose unit. While the intercalation of the nucleus into DNA and the groove binding of the sugar moiety provide potent topoisomerase I inhibition, the lack of adequate water solubility prevents effective pharmacological evaluation and formulation. Previous attempts to modify the structure often focused on the top amide group or the benzene rings, which yielded mixed results regarding solubility enhancement without compromising the critical DNA-binding architecture. Furthermore, the presence of chlorine atoms in the natural structure was found to have a negative impact on biological activity, necessitating a more refined structural optimization strategy that goes beyond simple halogen substitution.
The Novel Approach
The methodology outlined in CN102898489B represents a paradigm shift by targeting the N-6 nitrogen atom of the pyrrole ring for substitution. This site is strategically chosen because modifications here have minimal negative impact on the biological activity while offering a robust handle for attaching solubilizing groups. The patent describes four distinct series of analogues: N-glycosyl (Series I), N-amino acid (Series II), N-base (Series III), and N-alkyl/other substituents (Series IV). By introducing polar groups such as hydrophilic sugars, amino acids, and heterocyclic bases directly at this position, the resulting analogues exhibit dramatically improved water solubility. 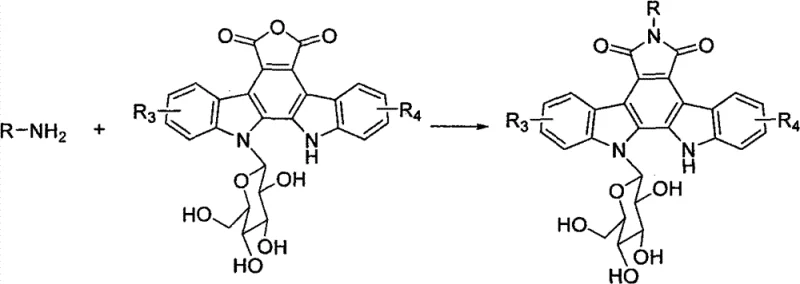 This approach preserves the essential indolocarbazole core required for DNA intercalation while leveraging the physicochemical benefits of the appended groups, effectively solving the solubility issue that has plagued previous iterations of this drug class.
This approach preserves the essential indolocarbazole core required for DNA intercalation while leveraging the physicochemical benefits of the appended groups, effectively solving the solubility issue that has plagued previous iterations of this drug class.
Mechanistic Insights into Glycosylation and Coupling Reactions
The synthesis of these advanced intermediates relies on precise control over glycosidic bond formation and nucleophilic substitution. For the N-glycosyl series, the process begins with the activation of sugar donors. Peracetylated sugars or azido-sugars are reacted with N-Phth aminoethanol in the presence of Lewis acids like boron trifluoride etherate (BF3·Et2O) or trimethylsilyl trifluoromethanesulfonate (TMSOTf). This step is critical for establishing the correct stereochemistry and linkage stability. The reaction conditions, often maintained at low temperatures such as -35°C to 0°C followed by warming to room temperature, ensure high selectivity and prevent degradation of the sensitive carbohydrate structures. Yields for these intermediate steps are reported to be robust, ranging from 50% to 99% depending on the specific sugar and protecting group strategy employed, indicating a highly efficient pathway for generating diverse glycosyl building blocks.
Following the preparation of the glycosyl intermediates, the core coupling reaction takes place. The key precursor, Compound 13 (the deprotected indolocarbazole core with a glucose unit), acts as the electrophilic partner or is coupled via nucleophilic attack depending on the specific series. In the case of Series I, the glycosyl intermediates (Compounds 7-12) are deprotected and then reacted with Compound 13 in dimethylformamide (DMF) at 80°C. 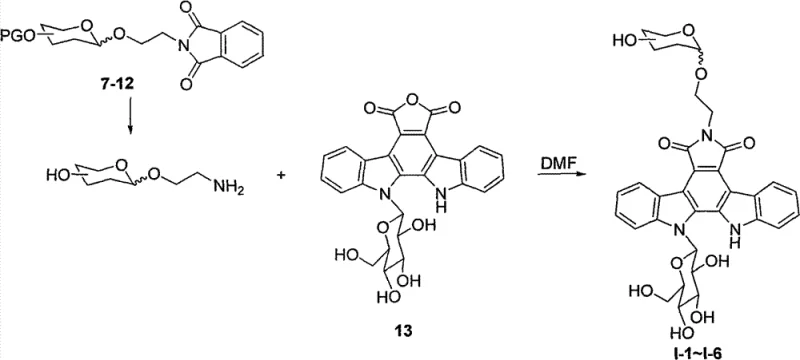 This thermal coupling facilitates the formation of the N-C or N-N bonds required to attach the new substituent. For amino acid and base analogues, the reaction involves heating Compound 13 with the respective amine or heterocycle in DMF at temperatures up to 150°C. The mechanism likely involves nucleophilic attack by the amine nitrogen on the activated core, displacing a leaving group or opening a cyclic anhydride intermediate. The high yields observed, such as 90% for certain amino acid derivatives and up to 98% for reduced azido-sugar analogues, demonstrate the reliability and reproducibility of this chemical transformation, which is essential for consistent batch-to-batch quality in pharmaceutical manufacturing.
This thermal coupling facilitates the formation of the N-C or N-N bonds required to attach the new substituent. For amino acid and base analogues, the reaction involves heating Compound 13 with the respective amine or heterocycle in DMF at temperatures up to 150°C. The mechanism likely involves nucleophilic attack by the amine nitrogen on the activated core, displacing a leaving group or opening a cyclic anhydride intermediate. The high yields observed, such as 90% for certain amino acid derivatives and up to 98% for reduced azido-sugar analogues, demonstrate the reliability and reproducibility of this chemical transformation, which is essential for consistent batch-to-batch quality in pharmaceutical manufacturing.
How to Synthesize N-6 Substituted Rebeccamycin Analogues Efficiently
The synthesis protocol described in the patent offers a modular approach that allows for the rapid generation of a library of analogues. The process generally involves three main stages: the preparation of the functionalized side chain (sugar, amino acid, or base), the synthesis of the indolocarbazole core precursor, and the final coupling step. Detailed standard operating procedures for each step, including specific molar ratios, solvent choices like dichloromethane and DMF, and purification methods such as column chromatography, are essential for replicating the high purity levels required for biological testing. The flexibility of this route allows chemists to swap out different R-groups easily, facilitating structure-activity relationship (SAR) studies to identify the most potent candidates.
- Prepare glycosyl donors by acetylating sugars or introducing azide groups using catalysts like ferric sulfate hydrate or copper sulfate.
- Synthesize glycosyl intermediates by reacting protected sugars with N-Phth aminoethanol using Lewis acids such as BF3·Et2O or TMSOTf.
- Couple the glycosyl intermediates with the deprotected indolocarbazole core (Compound 13) in DMF at elevated temperatures to form the final analogues.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the synthesis methods described in this patent offer significant advantages for supply chain stability and cost management in the production of anticancer intermediates. The reliance on readily available starting materials such as glucose, aminoethanol, and common heterocycles ensures that raw material sourcing is not a bottleneck. Unlike processes that require rare earth metals or exotic catalysts, this methodology utilizes standard organic reagents and Lewis acids that are accessible globally, reducing the risk of supply disruptions. Furthermore, the high yields reported in the experimental examples suggest a material-efficient process that minimizes waste generation, aligning with modern green chemistry principles and reducing disposal costs for manufacturing facilities.
- Cost Reduction in Manufacturing: The synthetic route is designed to be direct and efficient, avoiding unnecessary protection and deprotection steps where possible. For instance, the direct coupling of intermediates in DMF eliminates the need for complex multi-step sequences often seen in total synthesis. By streamlining the workflow, manufacturers can achieve substantial cost savings in terms of labor, solvent consumption, and reactor time. The elimination of transition metal catalysts in the final coupling steps also removes the need for expensive and rigorous heavy metal removal processes, which are a significant cost driver in API manufacturing. This streamlined approach translates to a more economically viable production model for high-value oncology intermediates.
- Enhanced Supply Chain Reliability: The robustness of the chemical reactions described, evidenced by consistent yields across different analogue series, ensures predictable production timelines. The use of stable intermediates that can be stored and transported without significant degradation adds another layer of security to the supply chain. Procurement managers can rely on the scalability of these reactions, as the conditions (e.g., reflux in DMF, stirring at room temperature) are easily transferable from laboratory glassware to large-scale industrial reactors. This scalability guarantees that as demand for these potent anticancer candidates grows during clinical trials, the supply can be ramped up seamlessly without requiring process re-engineering.
- Scalability and Environmental Compliance: The process generates byproducts that are manageable through standard wastewater treatment protocols, avoiding the creation of persistent organic pollutants. The high atom economy of the coupling reactions means less chemical waste per kilogram of product. Additionally, the ability to produce water-soluble analogues reduces the need for hazardous organic solvents in downstream formulation and testing phases. This environmental compatibility not only simplifies regulatory compliance but also enhances the corporate sustainability profile of the manufacturing entity, a key factor for partnerships with major pharmaceutical companies committed to ESG goals.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these Rebeccamycin analogues. Understanding these details is crucial for R&D teams evaluating the feasibility of incorporating these intermediates into their drug discovery pipelines. The answers are derived directly from the technical specifications and experimental data provided in the patent documentation, ensuring accuracy and relevance for decision-makers.
Q: What is the primary advantage of N-6 substituted Rebeccamycin analogues over the natural compound?
A: The primary advantage is significantly improved water solubility, which addresses the major limitation of natural Rebeccamycin and allows for better pharmacological evaluation and bioavailability in anticancer applications.
Q: What types of substituents can be introduced at the N-6 position?
A: A wide variety of hydrophilic groups can be introduced, including sugar moieties (glycosyl), amino acids, nucleic acid bases (like adenine and cytosine), and alkyl chains, allowing for fine-tuning of biological activity.
Q: Is this synthesis method scalable for commercial production?
A: Yes, the described methods utilize standard organic synthesis techniques and commercially available reagents, making the process adaptable for scale-up from laboratory to industrial manufacturing levels.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Rebeccamycin Analogues Supplier
NINGBO INNO PHARMCHEM stands at the forefront of custom synthesis for complex pharmaceutical intermediates, possessing the technical expertise to bring sophisticated molecules like N-6 substituted Rebeccamycin analogues from bench to bulk. Our team has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project needs are met with precision and speed. We maintain stringent purity specifications and operate rigorous QC labs equipped with advanced analytical instrumentation to guarantee that every batch meets the highest standards required for preclinical and clinical studies. Our commitment to quality assurance ensures that the impurities profile is tightly controlled, which is critical for the safety and efficacy of anticancer drug candidates.
We invite you to collaborate with us to leverage these innovative synthesis technologies for your oncology portfolio. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and route feasibility assessments to help you accelerate your development timeline. Let us be your partner in transforming promising chemical entities into life-saving medicines through superior manufacturing excellence and supply chain reliability.