Optimized Synthetic Route for Parecoxib Sodium Impurity: Enhancing Purity and Commercial Scalability
Optimized Synthetic Route for Parecoxib Sodium Impurity: Enhancing Purity and Commercial Scalability
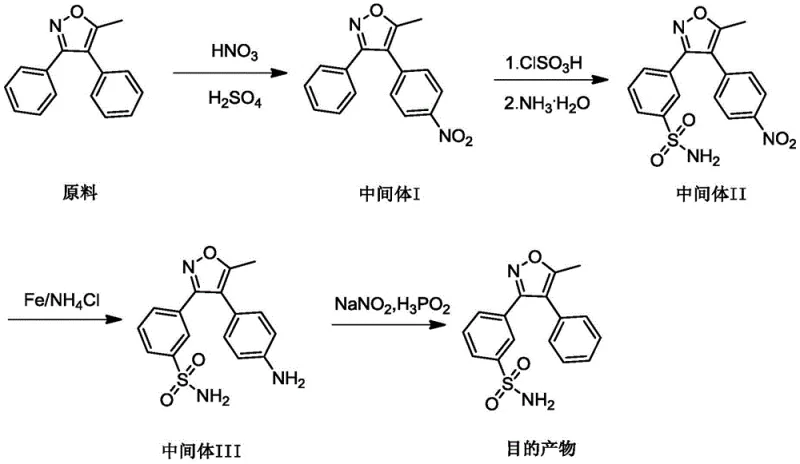
The pharmaceutical industry continuously demands higher standards for impurity profiling to ensure the safety and efficacy of active pharmaceutical ingredients (APIs). A critical development in this domain is documented in patent CN113773270A, which discloses a robust synthesis method for 3-(5-methyl-4-phenylisoxazol-3-yl)benzenesulfonamide, a key impurity associated with Parecoxib Sodium. This specific compound serves as an essential reference standard for quality control, enabling manufacturers to detect and quantify trace impurities that could affect patient safety. The disclosed methodology represents a significant advancement over existing techniques by streamlining the synthetic pathway, thereby reducing the cumulative loss of material often seen in multi-step processes. By optimizing reaction conditions such as temperature control and reagent stoichiometry, this approach ensures consistent batch-to-batch reproducibility, which is paramount for regulatory compliance in global markets. For R&D directors and procurement specialists, understanding the nuances of this patent provides a strategic advantage in sourcing high-quality intermediates that meet stringent pharmacopoeial requirements.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of complex sulfonamide intermediates like the Parecoxib Sodium impurity has been plagued by inefficient reaction pathways that compromise both yield and operational safety. Prior art methods, such as those cited in CN108047155, often suffer from competitive side reactions during the sulfonation step, leading to the formation of positional isomers that are difficult to separate. This necessitates the use of preparative liquid chromatography, a technique that is not only cost-prohibitive on a large scale but also significantly extends production lead times. Furthermore, alternative routes reported in literature frequently rely on hazardous reagents or require extreme reaction conditions that pose safety risks to personnel and equipment. The accumulation of these inefficiencies results in a final product with variable purity, making it unsuitable for use as a certified reference material in high-stakes drug quality research. Consequently, manufacturers face increased costs related to waste disposal, reprocessing, and the procurement of expensive purification media, creating a bottleneck in the supply chain for critical pharmaceutical intermediates.
The Novel Approach
In stark contrast to these traditional limitations, the method outlined in patent CN113773270A introduces a streamlined four-step sequence that prioritizes atom economy and operational simplicity. The process begins with the selective nitration of 5-methyl-3,4-diphenylisoxazole, followed by a highly efficient sulfonylation and amidation sequence that avoids the formation of difficult-to-remove isomers. By utilizing readily available reagents such as chlorosulfonic acid and ammonia water under controlled temperatures, the protocol minimizes the generation of hazardous byproducts. The subsequent reduction and diazotization steps are optimized to proceed under mild conditions, utilizing cost-effective reducing agents like iron powder or zinc powder instead of precious metal catalysts. This strategic selection of reagents not only lowers the raw material costs but also simplifies the downstream purification process, allowing for high-purity isolation through standard crystallization techniques. The result is a robust manufacturing route that delivers consistent quality while significantly reducing the environmental footprint associated with chemical synthesis.
Mechanistic Insights into Nitration and Diazotization Reduction
The core of this synthetic breakthrough lies in the precise control of electrophilic aromatic substitution and redox mechanisms throughout the reaction sequence. In the initial nitration step, the use of mixed acid (nitric and sulfuric acid) generates the nitronium ion, which selectively attacks the electron-rich phenyl ring of the isoxazole derivative. The patent specifies a molar ratio of raw material to nitrating agent between 1:1 and 1:5, ensuring complete conversion while minimizing over-nitration which could lead to dinitro byproducts. Maintaining the reaction temperature between 10-50°C is critical, as excessive heat can degrade the sensitive isoxazole ring structure, while insufficient heat slows the kinetics, leading to incomplete reactions. This careful balance ensures that Intermediate I is formed with high regioselectivity, setting the foundation for the subsequent sulfonation step where the sulfonyl group is introduced ortho to the nitro group. The mechanistic precision here is vital for R&D teams aiming to replicate the process, as slight deviations in acidity or temperature can drastically alter the impurity profile of the intermediate.
Furthermore, the final transformation involving diazotization and reduction demonstrates a sophisticated approach to removing the amino group to achieve the final unsubstituted phenyl structure. The conversion of the amine in Intermediate III to a diazonium salt using sodium nitrite at low temperatures (-10°C) prevents the decomposition of the unstable diazo species. The subsequent reduction using hypophosphorous acid acts as a radical scavenger, effectively replacing the diazo group with a hydrogen atom without affecting the sulfonamide or isoxazole moieties. This step is particularly crucial for impurity control, as incomplete reduction could leave behind azo-coupling byproducts that are structurally similar to the target molecule and difficult to separate. By optimizing the mass-volume ratio of the intermediate to the reducing agent (1:10 to 1:50 g/mL), the process ensures complete conversion, thereby maximizing the purity of the final 3-(5-methyl-4-phenylisoxazol-3-yl)benzenesulfonamide. This level of mechanistic control is what distinguishes this patent as a viable commercial process for high-purity reference standards.
How to Synthesize 3-(5-methyl-4-phenylisoxazol-3-yl)benzenesulfonamide Efficiently
Implementing this synthesis route requires strict adherence to the specified reaction parameters to ensure safety and yield optimization. The process is designed to be scalable, moving from laboratory glassware to industrial reactors with minimal modification to the core chemistry. Operators must focus on the precise addition rates of chlorosulfonic acid during the sulfonylation step, as this exothermic reaction requires careful thermal management to prevent runaway scenarios. Additionally, the workup procedures, particularly the extraction and crystallization stages, are critical for removing inorganic salts and residual solvents that could impact the final purity specifications. The following guide outlines the standardized operational steps derived from the patent examples, serving as a foundational protocol for process chemists.
- Perform nitration of 5-methyl-3,4-diphenylisoxazole using nitric acid or mixed acid at 10-50°C to obtain Intermediate I.
- Execute sulfonylation with chlorosulfonic acid followed by amidation with ammonia water to generate Intermediate II.
- Conduct nitro reduction using iron powder or zinc powder in ethanol/water to yield Intermediate III.
- Finalize synthesis via diazotization with sodium nitrite and reduction with hypophosphorous acid to obtain the target sulfonamide.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this synthetic route offers tangible benefits that extend beyond mere chemical efficiency. The primary advantage lies in the substantial cost reduction in pharmaceutical intermediates manufacturing achieved through the elimination of expensive catalysts and complex purification steps. By avoiding the use of precious metals like palladium in the reduction phase and opting for iron or zinc powder, the raw material costs are drastically lowered without compromising reaction efficiency. Furthermore, the ability to perform sulfonylation under solvent-free conditions or with minimal solvent usage reduces the volume of hazardous waste generated, leading to significant savings in waste treatment and disposal fees. These factors combine to create a more economical production model that allows suppliers to offer competitive pricing while maintaining healthy margins, a critical consideration for long-term supply contracts in the volatile chemical market.
- Cost Reduction in Manufacturing: The strategic selection of reagents directly impacts the bottom line by removing the need for costly transition metal catalysts and extensive chromatographic purification. Traditional methods often require preparative HPLC to separate isomers, a process that consumes large volumes of solvents and stationary phases, driving up operational expenses. In contrast, this novel approach relies on crystallization and standard extraction, which are inherently cheaper and faster. The reduction in step count also means less labor time and lower energy consumption per kilogram of product, contributing to overall operational efficiency. Consequently, manufacturers can achieve a more favorable cost structure, enabling them to absorb fluctuations in raw material prices while remaining competitive in the global marketplace.
- Enhanced Supply Chain Reliability: The reliance on commodity chemicals such as nitric acid, sulfuric acid, and iron powder ensures that the supply chain is resilient against disruptions. Unlike specialized reagents that may have limited suppliers or long lead times, the materials required for this synthesis are widely available from multiple global sources. This diversification of supply reduces the risk of production halts due to raw material shortages, ensuring consistent delivery schedules for downstream pharmaceutical clients. Additionally, the robustness of the reaction conditions means that the process is less sensitive to minor variations in reagent quality, further stabilizing production output. For supply chain heads, this translates to reduced safety stock requirements and improved inventory turnover rates.
- Scalability and Environmental Compliance: The process is inherently designed for scale-up, with reaction conditions that are easily manageable in large-scale reactors. The avoidance of high-pressure hydrogenation or cryogenic conditions simplifies the engineering requirements for production facilities, lowering capital expenditure for new capacity. Moreover, the reduced solvent usage and the ability to recycle aqueous streams align with increasingly stringent environmental regulations. By minimizing the generation of hazardous waste and volatile organic compounds (VOCs), manufacturers can maintain compliance with environmental standards without incurring excessive mitigation costs. This sustainability aspect is becoming a key differentiator in supplier selection, as pharmaceutical companies prioritize partners with strong environmental, social, and governance (ESG) credentials.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of this Parecoxib Sodium impurity. These answers are derived directly from the technical specifications and beneficial effects described in the patent documentation, providing clarity for potential partners. Understanding these details is essential for evaluating the feasibility of integrating this intermediate into your quality control workflows or production pipelines.
Q: What is the primary advantage of this synthesis method over prior art?
A: This method significantly reduces reaction steps and avoids the use of expensive or difficult-to-obtain reagents, resulting in higher overall yield and safer operation conditions compared to traditional multi-step pathways.
Q: What purity levels can be achieved with this protocol?
A: The process is designed to produce high-purity reference substances suitable for drug quality research, with HPLC purity data demonstrating levels exceeding 92% in validated examples, ensuring reliability for analytical standards.
Q: Is this process scalable for commercial intermediate production?
A: Yes, the use of common reagents like iron powder and chlorosulfonic acid, along with solvent-free options for sulfonylation, indicates strong potential for commercial scale-up without requiring specialized high-pressure equipment.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3-(5-methyl-4-phenylisoxazol-3-yl)benzenesulfonamide Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-purity impurities play in ensuring the safety and efficacy of pharmaceutical products. Our team of expert process chemists has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the synthetic route described in patent CN113773270A can be effectively translated into industrial reality. We maintain stringent purity specifications and operate rigorous QC labs equipped with advanced analytical instrumentation to verify every batch against the highest industry standards. Our commitment to quality assurance means that every gram of 3-(5-methyl-4-phenylisoxazol-3-yl)benzenesulfonamide we supply is accompanied by comprehensive documentation, including detailed COA data and stability profiles, giving you confidence in your drug quality research.
We invite you to collaborate with us to optimize your supply chain for this critical intermediate. By leveraging our technical expertise, you can secure a stable source of high-quality material that supports your regulatory filings and production needs. Please contact our technical procurement team to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our manufacturing capabilities can enhance your operational efficiency and reduce overall procurement costs.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →