Optimizing 5-HT4 Agonist Intermediate Production for Commercial Scale-Up
The pharmaceutical industry's relentless pursuit of effective treatments for neurodegenerative disorders has placed significant focus on the 5-HT4 receptor agonists, which show immense potential in addressing conditions such as Alzheimer's disease and schizophrenia. Central to the development of these therapeutic agents is the efficient production of key chemical building blocks, specifically 4-amino-5-halogenobenzofuran-7-carboxylic acid. Recent advancements documented in patent CN110818661A have introduced a groundbreaking preparation method that fundamentally alters the manufacturing landscape for this critical intermediate. This innovation not only simplifies the synthetic pathway but also dramatically enhances the overall economic viability of producing high-purity pharmaceutical intermediates. By leveraging a streamlined three-step reaction sequence, this technology addresses the long-standing challenges of low yield and complex purification that have historically plagued the supply chain for 5-HT4 receptor ligands. For R&D directors and procurement managers alike, understanding the technical nuances of this patent is essential for securing a reliable pharmaceutical intermediates supplier capable of meeting the rigorous demands of modern drug development.
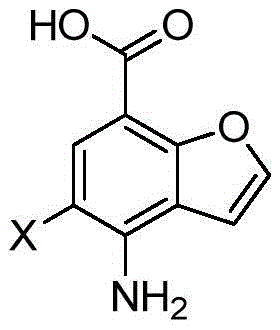
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior to the innovations disclosed in CN110818661A, the synthesis of 4-amino-5-halogenobenzofuran-7-carboxylic acid was predominantly governed by cumbersome multi-step protocols, such as the route detailed in WO 2016128990A1. This legacy approach necessitates a tedious six-step sequence involving esterification, acetyl protection, chlorination, iodination, Sonogashira coupling, and final ring closure. Each additional step in this linear synthesis introduces inherent risks of material loss, accumulation of impurities, and significant increases in operational costs. The reliance on expensive iodination reagents and the need for rigorous purification after every transformation result in a dismal external standard yield of merely 24% when repeated on a gram scale. Furthermore, the amplification of this process to the kilogram scale often exacerbates these inefficiencies, dropping yields even further to around 22%. Such poor economic benefits and difficult purification processes create substantial bottlenecks for commercial scale-up of complex pharmaceutical intermediates, making the conventional method unsustainable for large-volume production requirements.
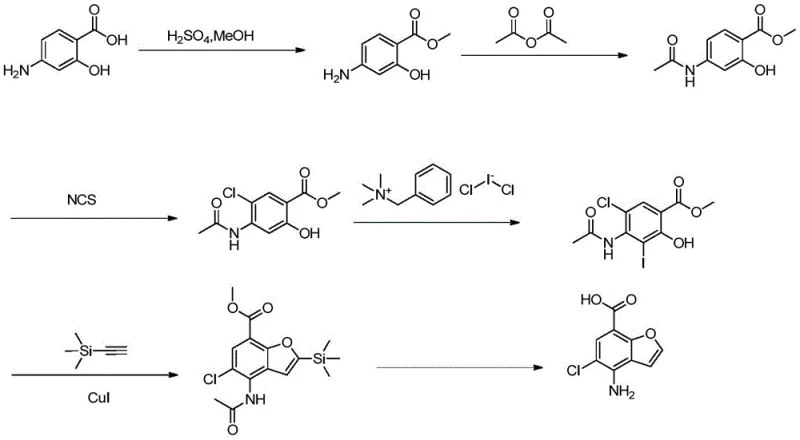
The Novel Approach
In stark contrast to the convoluted legacy pathways, the novel approach presented in the patent data offers a remarkably concise and robust three-step synthesis that bypasses the need for excessive protection and deprotection cycles. This method utilizes para-protected amino-o-hydroxybenzoic acid esters and triethylacetylene silicon as main raw materials, reacting them through a direct halogenation followed by a catalytic cyclization. By generating two halogen atoms at the 3 and 5 positions simultaneously during the halogenation step, the process effectively simplifies the reaction operation steps and eliminates the need for separate iodination procedures. The result is a total yield exceeding 51%, which represents a more than twofold improvement over the prior art. This drastic simplification not only reduces the consumption of solvents and reagents but also significantly shortens the production cycle time. For supply chain heads, this translates to a more predictable and efficient manufacturing timeline, ensuring that cost reduction in API manufacturing is achieved through fundamental process intensification rather than mere negotiation.
Mechanistic Insights into Pd-Catalyzed Cyclization and Halogenation
The core of this technological breakthrough lies in the precise control of regioselectivity during the halogenation and the subsequent palladium-catalyzed cyclization. In the first step, the reaction of 4-protected amino-2-hydroxybenzoic acid esters with halogenating reagents like N-chlorosuccinimide or N-bromosuccinimide is carefully optimized to ensure dihalogenation occurs specifically at the 3 and 5 positions. This selectivity is crucial as it pre-organizes the molecule for the subsequent ring-closing event, avoiding the formation of isomeric byproducts that would complicate downstream purification. The use of solvents such as 1,2-dichloroethane facilitates this transformation by dissolving the reactants effectively without interfering with the reaction yield. Following this, the intermediate undergoes a coupling reaction with trialkyl acetylene silicon in the presence of a palladium catalyst and cuprous iodide. This step is the mechanistic heart of the synthesis, where the alkyne moiety is introduced and subsequently cyclized to form the benzofuran core.
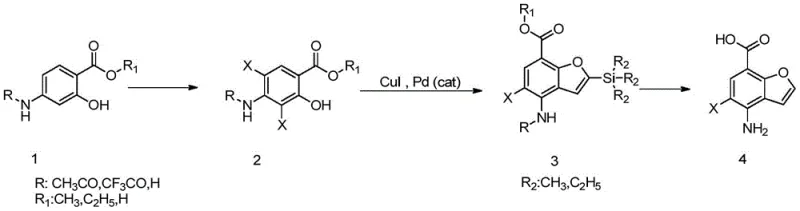
Impurity control is inherently built into this mechanism through the stability of the intermediates and the specificity of the catalysts employed. The palladium catalyst, selected from options like bis(triphenylphosphine)palladium dichloride, ensures high turnover and minimizes the formation of homocoupling byproducts often seen in Sonogashira-type reactions. Furthermore, the final deprotection and hydrolysis step under alkaline conditions is designed to be clean and quantitative, removing silyl and acetyl groups without degrading the sensitive benzofuran ring. The ability to complete the ring-closing reaction efficiently when the 3-position is chlorine or bromine allows for the generation of the final halogenated product in a single pot during the coupling phase. This mechanistic elegance ensures that the impurity profile of the final high-purity OLED material or pharmaceutical intermediate remains within stringent specifications, reducing the burden on quality control laboratories and ensuring batch-to-batch consistency essential for regulatory compliance.
How to Synthesize 4-Amino-5-Halogenobenzofuran-7-Carboxylic Acid Efficiently
Implementing this synthesis route requires careful attention to reaction conditions and reagent stoichiometry to maximize the benefits of the patented process. The procedure begins with the halogenation of the protected starting material, followed by the critical palladium-catalyzed coupling, and concludes with a straightforward hydrolysis. Each stage has been optimized to balance reaction rate with product quality, ensuring that the process is not only theoretically sound but practically viable for industrial application. The detailed standardized synthesis steps below outline the specific parameters required to replicate the high yields reported in the patent data, providing a clear roadmap for technical teams looking to adopt this methodology.
- Halogenation of 4-protected amino-2-hydroxybenzoic acid ester using N-halosuccinimide to form the dihalo intermediate.
- Pd/Cu-catalyzed coupling with trialkylacetylene silicon to induce cyclization and form the benzofuran core.
- Alkaline deprotection and hydrolysis to remove protecting groups and yield the final carboxylic acid product.
Commercial Advantages for Procurement and Supply Chain Teams
The transition from a six-step legacy process to this streamlined three-step methodology offers profound commercial advantages that extend far beyond simple yield improvements. For procurement managers, the reduction in reaction steps directly correlates to a significant decrease in the consumption of raw materials, solvents, and energy, leading to substantial cost savings in the overall manufacturing budget. The elimination of expensive iodination reagents and the reduction in purification stages mean that the cost of goods sold (COGS) can be drastically optimized without compromising on the quality of the final product. This efficiency makes the production of high-purity pharmaceutical intermediates more economically sustainable, allowing for competitive pricing strategies in the global market. Moreover, the use of cheap and easily obtained raw materials enhances supply chain resilience, reducing the risk of disruptions caused by the scarcity of specialized reagents.
- Cost Reduction in Manufacturing: The primary driver for cost reduction lies in the minimization of unit operations. By cutting the synthesis from six steps to three, the process eliminates three distinct isolation and purification events, each of which typically incurs significant labor, equipment, and material costs. The removal of the iodination step specifically targets a high-cost variable, replacing it with more economical chlorination or bromination reagents that are readily available in bulk quantities. Additionally, the higher overall yield means that less starting material is required to produce the same amount of final product, effectively amplifying the purchasing power of the procurement budget. This qualitative improvement in process efficiency ensures that cost reduction in API manufacturing is achieved through structural optimization rather than temporary market fluctuations.
- Enhanced Supply Chain Reliability: Supply chain reliability is significantly bolstered by the simplicity and robustness of the new reaction pathway. Fewer reaction steps inherently reduce the probability of batch failures, as there are fewer opportunities for human error or equipment malfunction to compromise the product. The use of common organic solvents and stable catalysts further ensures that the process is not dependent on fragile or hard-to-source chemicals that could lead to lead time delays. This stability allows for more accurate production planning and inventory management, ensuring that reducing lead time for high-purity pharmaceutical intermediates becomes a tangible reality. Consequently, partners can rely on a consistent flow of materials, supporting continuous manufacturing operations without the fear of unexpected stoppages.
- Scalability and Environmental Compliance: Scalability is a critical factor for any process intended for commercial production, and this method has demonstrated excellent potential for amplification from gram to kilogram scales. The simple operation conditions, such as moderate heating temperatures and standard atmospheric pressure, make it easy to transfer the process from the laboratory to large-scale reactors without requiring specialized high-pressure equipment. Furthermore, the reduction in solvent usage and waste generation aligns with increasingly strict environmental regulations, simplifying the handling of three wastes and lowering disposal costs. The ability to smoothly amplify to the kilogram-level reaction scale while maintaining high purity ensures that the process is ready for immediate industrial mass production, meeting the demands of a growing market for 5-HT4 receptor agonists.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis route. They are derived from the specific technical advantages and operational details found within the patent documentation, providing clarity for stakeholders evaluating the feasibility of this technology. Understanding these aspects is crucial for making informed decisions about process adoption and supply chain integration.
Q: How does the new three-step route compare to previous six-step methods in terms of yield?
A: The novel three-step process described in patent CN110818661A achieves a total yield of over 51%, significantly outperforming the conventional six-step route which typically yields only around 24%.
Q: What are the primary cost drivers eliminated in this new synthesis method?
A: By reducing the reaction steps from six to three, the process eliminates multiple isolation and purification stages, reduces solvent consumption, and avoids the use of expensive iodination reagents required in the older pathway.
Q: Is this process suitable for large-scale industrial manufacturing?
A: Yes, the method utilizes cheap and easily obtained raw materials, features simple operation conditions, and has demonstrated successful amplification to kilogram-scale reactions with consistent reproducibility.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 4-Amino-5-Halogenobenzofuran-7-Carboxylic Acid Supplier
The technical potential of this three-step synthesis route represents a significant opportunity for pharmaceutical companies looking to optimize their supply chains for 5-HT4 receptor agonists. NINGBO INNO PHARMCHEM stands ready as a premier CDMO partner, possessing extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our team of experts is well-versed in the nuances of palladium-catalyzed reactions and halogenation chemistry, ensuring that the transition from patent to production is seamless and efficient. We maintain stringent purity specifications and operate rigorous QC labs to guarantee that every batch of 4-amino-5-halogenobenzofuran-7-carboxylic acid meets the highest industry standards. Our commitment to quality and reliability makes us the ideal choice for organizations seeking a reliable pharmaceutical intermediates supplier who can deliver both technical excellence and commercial value.
We invite you to engage with our technical procurement team to discuss how this innovative synthesis route can be tailored to your specific production needs. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into the potential economic benefits of adopting this method for your specific application. We encourage you to reach out for specific COA data and route feasibility assessments to verify the compatibility of this process with your existing manufacturing infrastructure. Together, we can drive forward the development of next-generation therapeutics with a supply chain that is both robust and cost-effective.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →