Advanced Purification Technology for Azasetron Hydrochloride: Ensuring High Purity and Commercial Scalability
The pharmaceutical landscape for antiemetic agents continues to demand higher standards of purity and safety, particularly for potent serotonin 5-HT3 receptor antagonists like Azasetron Hydrochloride. Patent CN102329313B introduces a transformative purification methodology that addresses the chronic issues of polymeric impurities and heavy metal contamination found in conventionally synthesized batches. This technical breakthrough is not merely a laboratory curiosity but a robust industrial solution designed to elevate the quality profile of Y-25130 bulk drug substances. By implementing a strategic sequence of solvent washing, pH-controlled phase transfer, and precision crystallization, manufacturers can now achieve purity levels exceeding 99.5%, thereby mitigating the toxicological risks associated with long-term administration. For R&D directors and procurement specialists alike, this patent represents a pivotal shift towards domestic self-sufficiency and superior quality control in the supply chain of critical oncology support medications.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthetic routes for Azasetron Hydrochloride often suffer from significant downstream processing challenges that compromise the final quality of the active pharmaceutical ingredient. Standard protocols typically involve harsh reaction conditions that promote the formation of stubborn polymeric byproducts and leave behind trace amounts of heavy metal catalysts used in earlier coupling steps. These impurities are notoriously difficult to remove using simple recrystallization techniques because they often co-crystallize with the target molecule or remain trapped within the crystal lattice. Furthermore, conventional drying processes at elevated temperatures can exacerbate degradation, leading to increased coloration and a decline in assay potency. The reliance on imported raw materials to bypass these domestic quality deficits has historically created supply chain vulnerabilities, forcing manufacturers to accept lower yields and higher costs while struggling to meet stringent international pharmacopeia standards for related substances and residual solvents.
The Novel Approach
The innovative purification strategy outlined in the patent data fundamentally re-engineers the isolation process to prioritize impurity rejection over simple recovery. Instead of attempting to purify the final salt directly from a complex reaction mixture, this method employs a clever 'dissolve-and-precipitate' logic that exploits the differential solubility of the free base versus its hydrochloride salt. By first washing the crude material with specific insoluble solvents at controlled low temperatures, the process effectively strips away organic polymeric contaminants before the main purification even begins. Subsequent treatment with ammonia water converts the active ingredient into its free base form, which remains soluble in the aqueous phase while forcing inorganic salts and heavy metals to precipitate out as insoluble sludge. This phase separation is the key differentiator, allowing for a level of cleanliness that traditional acid-base workups simply cannot achieve without expensive chromatographic intervention.
Mechanistic Insights into Acid-Base Extraction and Controlled Crystallization
The core chemical mechanism driving this purification success lies in the precise manipulation of protonation states and solubility equilibria. In the initial stage, the use of solvents like ethanol or ethyl acetate at temperatures strictly below 30°C ensures that the target Azasetron Hydrochloride remains in the solid phase while highly soluble organic impurities are washed away. This step is critical because polymeric impurities, often formed during the amidation or cyclization stages of synthesis, possess different polarity profiles that make them susceptible to removal by these specific organic washes. Following this, the introduction of ammoniacal liquor raises the pH to a weakly alkaline range (pH ≤ 9), deprotonating the amine nitrogen on the azabicyclooctane ring. This conversion to the neutral free base increases its lipophilicity and solubility in the aqueous-ammonia matrix, whereas metal ions and inorganic anions form insoluble hydroxides or salts that are mechanically removed via filtration, effectively acting as a chemical scavenger step without the need for added resins.
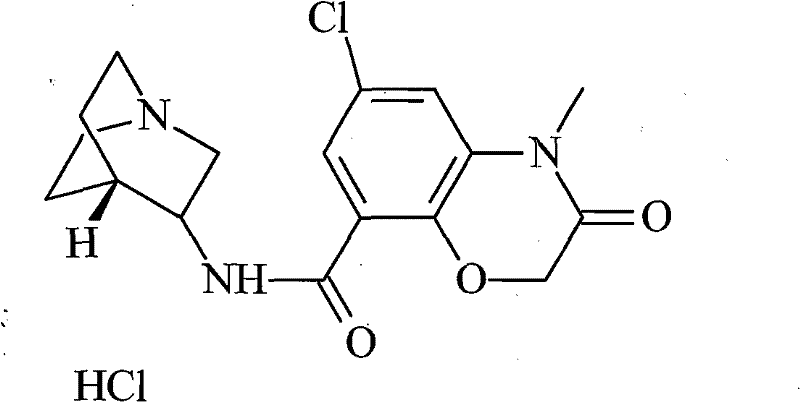
The final crystallization step is a masterclass in thermodynamic control, where the reintroduction of hydrochloric acid into an ethanol-water mixture triggers the reformation of the hydrochloride salt under highly regulated conditions. By maintaining the temperature between 30-60°C and carefully titrating the pH to a narrow window of 0.5-3.0, the process encourages the growth of large, well-defined crystals rather than the rapid precipitation of amorphous powder. Slow crystal growth is essential for purity because it allows the crystal lattice to reject impurity molecules that do not fit sterically or electronically, a phenomenon known as zone refining during solidification. The gradual cooling to a minimum of 10°C further maximizes yield without trapping mother liquor inclusions, ensuring that the final dried product exhibits excellent flow properties and minimal solvent retention, which are critical parameters for downstream tablet compression or capsule filling operations.
How to Synthesize Azasetron Hydrochloride Efficiently
Implementing this purification protocol requires strict adherence to temperature and pH parameters to replicate the high-purity results demonstrated in the patent examples. The process transforms low-grade bulk drug or crude synthesis output into a premium-grade intermediate suitable for direct formulation. Operators must focus on the gentle handling of the free base intermediate to prevent oxidation and ensure complete removal of inorganic precipitates before the final acidification step. The detailed standardized synthesis steps, including specific solvent ratios and agitation speeds required for optimal crystal habit formation, are outlined in the technical guide below for immediate implementation by process engineering teams.
- Wash crude Azasetron HCl with insoluble solvents like ethanol or ethyl acetate at temperatures below 30°C to remove organic impurities and polymers.
- Treat the filter cake with ammonia water (pH ≤ 9) to convert the salt to its free base form, dissolving the active compound while precipitating inorganic salts and heavy metals.
- Add ethanol and drip concentrated hydrochloric acid into the solution at 30-60°C, controlling pH between 0.5-3.0 to induce slow crystallization of high-purity Azasetron Hydrochloride.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this advanced purification technology offers substantial strategic benefits beyond mere technical compliance. By enabling the production of high-purity Azasetron Hydrochloride from domestically sourced crude materials, pharmaceutical companies can drastically reduce their dependency on volatile import markets and mitigate currency exchange risks. The elimination of complex chromatographic purification steps translates directly into simplified manufacturing workflows, reduced consumption of expensive stationary phases, and lower waste disposal costs associated with solvent-heavy processes. Furthermore, the robustness of this crystallization-based method ensures consistent batch-to-batch quality, which minimizes the risk of regulatory rejection and costly re-processing, thereby stabilizing the overall cost of goods sold and enhancing the reliability of supply for critical antiemetic medications.
- Cost Reduction in Manufacturing: The process achieves significant cost optimization by replacing expensive and time-consuming column chromatography with simple filtration and crystallization unit operations. By effectively removing heavy metals and polymeric impurities through chemical means rather than physical separation on resin, the method eliminates the need for costly scavenger resins and reduces solvent consumption volumes. This streamlined approach lowers the operational expenditure per kilogram of produced API, allowing for more competitive pricing in the generic pharmaceutical market while maintaining healthy profit margins through improved yield efficiency and reduced waste treatment burdens.
- Enhanced Supply Chain Reliability: Adopting this purification method empowers manufacturers to utilize a broader range of crude starting materials, including those with slightly lower initial purity, without compromising the final product quality. This flexibility reduces the risk of production stoppages caused by the unavailability of ultra-high-grade raw materials and shortens the lead time for raw material procurement. Additionally, the scalability of the process ensures that supply can be rapidly ramped up to meet sudden spikes in demand, such as during flu seasons or expanded oncology treatment protocols, providing a secure and resilient supply chain backbone for finished dosage form manufacturers.
- Scalability and Environmental Compliance: The purification route is inherently green and scalable, utilizing common solvents like ethanol and ethyl acetate which are easier to recover and recycle compared to exotic chlorinated solvents. The reduction in heavy metal content in the final product simplifies wastewater treatment requirements, as the metals are removed early in the process as solid sludge rather than remaining in liquid effluent streams. This alignment with environmental, social, and governance (ESG) goals not only reduces regulatory compliance costs but also enhances the corporate sustainability profile, making the supply chain more attractive to environmentally conscious global partners and investors.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this purification technology. They are derived from the specific constraints and advantages detailed in the patent documentation, focusing on the practical realities of scaling this chemistry from the bench to the plant floor. Understanding these nuances is essential for technical teams evaluating the feasibility of integrating this process into existing manufacturing lines.
Q: How does this purification method reduce polymeric impurities?
A: The process utilizes a specific solvent wash at low temperatures (<30°C) which selectively dissolves organic polymeric byproducts formed during previous synthesis steps without dissolving the target Azasetron Hydrochloride, effectively separating them before the final crystallization.
Q: Why is the ammonia treatment step critical for quality?
A: Converting the hydrochloride salt to the free base in an alkaline environment allows inorganic impurities, such as heavy metal catalysts and inorganic salts, to precipitate out as insoluble hydroxides or salts, which are then removed by filtration, significantly lowering residue on ignition.
Q: Can this process be scaled for industrial production?
A: Yes, the method relies on standard unit operations such as filtration, pH adjustment, and temperature-controlled crystallization, which are easily scalable from laboratory benchtop to multi-ton reactor systems without requiring complex chromatography equipment.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Azasetron Hydrochloride Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition from patent theory to commercial reality requires deep process engineering expertise and unwavering commitment to quality. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the delicate balance of temperature and pH described in the purification protocol is maintained even in large-scale reactors. We operate stringent purity specifications and utilize rigorous QC labs equipped with state-of-the-art HPLC and GC systems to verify that every batch of Azasetron Hydrochloride meets the >99.5% purity benchmark, guaranteeing that your final formulations are safe, effective, and compliant with global regulatory standards.
We invite you to collaborate with us to optimize your supply chain and reduce your manufacturing costs through the adoption of this superior purification technology. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating exactly how switching to our refined grade intermediates can improve your overall operational efficiency. Please contact us today to request specific COA data and route feasibility assessments, and let us help you secure a stable, high-quality supply of this critical antiemetic intermediate for your global markets.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →