Advanced Synthetic Route for Docetaxel Impurity A Ensuring High Purity and Scalability
Advanced Synthetic Route for Docetaxel Impurity A Ensuring High Purity and Scalability
The pharmaceutical industry faces increasing regulatory pressure to characterize and control impurities in potent oncology drugs like Docetaxel. Patent CN115650882A introduces a groundbreaking methodology for the preparation of a specific Docetaxel impurity, designated as Compound A, which is critical for establishing robust quality control standards. This novel synthetic pathway addresses the historical scarcity of authentic reference materials, enabling more precise analytical validation. By leveraging a sophisticated 7-step sequence involving orthogonal protection strategies and mild reaction conditions, this technology offers a reliable solution for producing high-purity intermediates. For R&D directors and procurement specialists, understanding this route is essential for securing a stable supply of critical quality attributes (CQAs) materials.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of complex taxane-related impurities has been plagued by significant technical hurdles, primarily due to the sensitivity of the molecular scaffold. Conventional approaches often rely on harsh acidic or basic conditions that can induce epimerization or degradation of the chiral centers, leading to complex mixtures that are difficult to separate. Furthermore, many existing literature methods lack a systematic protection strategy, resulting in poor regioselectivity during functionalization steps. This often necessitates extensive and costly purification procedures, such as repetitive preparative HPLC, which drastically reduces overall throughput. The absence of a standardized, high-yielding protocol has previously forced manufacturers to rely on inconsistent sources, compromising the reliability of impurity profiling in final drug products.
The Novel Approach
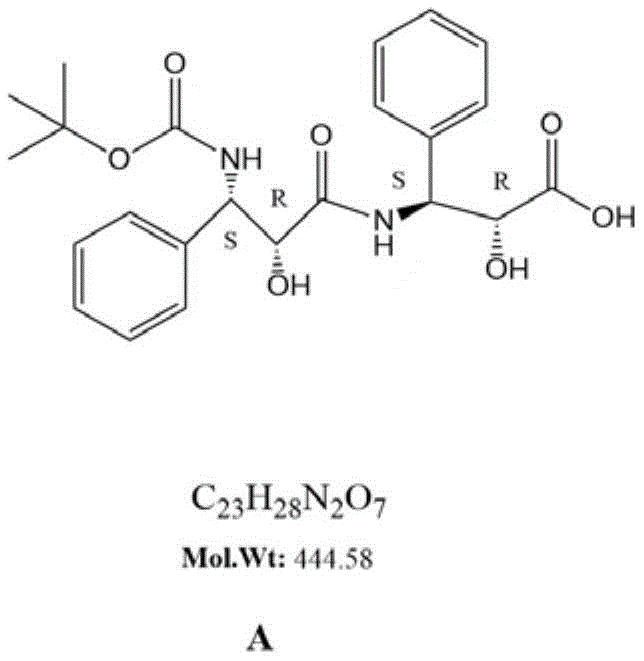
The methodology disclosed in patent CN115650882A represents a paradigm shift by employing a dual-protection strategy using tert-butyloxycarbonyl (Boc) and benzyloxycarbonyl (Cbz) groups. This orthogonal approach allows chemists to selectively manipulate the amine and hydroxyl functionalities independently, preventing unwanted side reactions. The process initiates with the protection of the substrate A-1, followed by a controlled SN2 methylation and subsequent amidation. Crucially, the reaction conditions are maintained within a mild temperature range, typically between -5°C and 0°C, which preserves the stereochemical integrity of the molecule. This precision engineering of the synthetic route ensures that the final product, Compound A, is obtained with superior purity, directly addressing the pain points of traditional synthesis.
Mechanistic Insights into Orthogonal Protection and Coupling Strategy
The core of this synthesis lies in the meticulous management of reactive functional groups through orthogonal protection. Initially, the amino group of the starting material, Phenylserine derivative A-1, is masked using di-tert-butyl dicarbonate (Boc2O) in an alkaline environment. This step is critical as it prevents the amine from interfering during the subsequent esterification and alkylation phases. Following this, the hydroxyl group is protected with benzyl chloroformate (Cbz-Cl), creating a fully protected intermediate A-3. The stability of the Cbz group allows for the subsequent SN2 reaction with methyl iodide to proceed efficiently, introducing the necessary methyl group without disturbing the nitrogen center. This sequential protection creates a robust scaffold that can withstand the rigors of subsequent transformations.
Following the construction of the methylated fragment, the strategy shifts to deprotection and coupling. The Boc group is selectively removed using trifluoroacetic acid (TFA), regenerating the free amine in intermediate A-5 while leaving the Cbz group intact. This free amine is then coupled with another equivalent of the protected acid fragment A-3 using modern peptide coupling reagents like PyBOP and HOBt. This amidation step forms the central peptide bond of the impurity structure. Finally, catalytic hydrogenolysis removes the Cbz group, and a concluding hydrolysis step reveals the carboxylic acid, yielding the target Docetaxel Impurity A. This mechanistic elegance ensures high fidelity in the final structure.
How to Synthesize Docetaxel Impurity A Efficiently
The execution of this synthesis requires precise control over stoichiometry and temperature to maximize yield and minimize impurities. The process begins with the dissolution of substrate A-1 in tetrahydrofuran and aqueous sodium hydroxide, cooled to -5-0°C before the slow addition of Boc2O. Subsequent steps involve careful pH adjustments during workup to ensure optimal phase separation. The amidation step, in particular, benefits from the use of anhydrous dichloromethane and inert atmosphere conditions to prevent hydrolysis of the activated ester. For a comprehensive, step-by-step laboratory protocol including exact reagent grades and safety precautions, please refer to the detailed guide below.
- Protect the amino group of substrate A-1 with di-tert-butyl dicarbonate (Boc2O) under alkaline conditions to form intermediate A-2.
- Protect the hydroxyl group of A-2 with benzyl chloroformate (Cbz-Cl) to yield A-3, followed by SN2 methylation using methyl iodide to generate A-4.
- Remove the Boc protecting group from A-4 using trifluoroacetic acid to obtain A-5, then couple A-5 with A-3 using PyBOP/HOBt condensing agents.
- Perform catalytic hydrogenolysis on the coupled product A-6 to remove the Cbz group, followed by ester hydrolysis to yield the final Impurity A.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented synthesis route offers tangible strategic benefits beyond mere technical feasibility. The reliance on commodity reagents such as Boc2O, Cbz-Cl, and methyl iodide ensures that raw material sourcing is not a bottleneck, as these chemicals are globally available from multiple vendors. Furthermore, the mild reaction temperatures eliminate the need for specialized cryogenic equipment capable of reaching extreme lows, thereby reducing capital expenditure and energy costs associated with cooling. The robustness of the purification steps, which utilize standard solvent systems like ethyl acetate and petroleum ether, simplifies waste management and solvent recovery processes.
- Cost Reduction in Manufacturing: The elimination of expensive transition metal catalysts and the use of ambient or near-ambient pressure hydrogenation significantly lowers the operational cost profile. By avoiding complex enzymatic resolutions or chiral chromatography in the early stages, the process achieves cost efficiency through chemical selectivity rather than expensive physical separation. The high atom economy of the coupling steps further contributes to reduced raw material consumption per kilogram of final product, driving down the overall cost of goods sold (COGS) for this critical reference standard.
- Enhanced Supply Chain Reliability: The modular nature of this 7-step synthesis allows for flexible production scheduling. Intermediates such as A-3 and A-5 can be stockpiled as stable inventory, decoupling the production timeline and ensuring rapid response to urgent demand spikes. Since the route does not depend on scarce natural extracts or single-source biological enzymes, the risk of supply disruption is minimized. This resilience is crucial for maintaining continuous quality control operations in large-scale API manufacturing facilities where downtime is not an option.
- Scalability and Environmental Compliance: The process is inherently scalable, transitioning smoothly from gram-scale laboratory synthesis to multi-kilogram production without fundamental changes to the chemistry. The solvents used are predominantly Class 3 or low-risk Class 2 solvents, facilitating easier regulatory compliance and environmental permitting. Additionally, the high purity achieved reduces the burden on downstream waste treatment facilities, as fewer toxic by-products are generated. This alignment with green chemistry principles enhances the corporate sustainability profile of the manufacturing partner.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of this Docetaxel impurity. These insights are derived directly from the experimental data and beneficial effects reported in the patent documentation, providing clarity for technical decision-makers. Understanding these nuances is vital for integrating this material into your quality assurance workflows effectively.
Q: Why is the synthesis of this specific Docetaxel impurity critical for API quality control?
A: As Docetaxel is a potent anti-tumor agent, regulatory bodies require rigorous identification and quantification of all related substances. This specific impurity, structurally characterized as a dimer-like derivative, can arise during semi-synthetic processes. Having an authentic reference standard synthesized via the patented CN115650882A route allows quality control laboratories to accurately validate analytical methods, ensuring the safety and efficacy of the final drug product.
Q: What are the key advantages of this 7-step synthesis compared to traditional methods?
A: The patented method utilizes a strategic orthogonal protection strategy involving Boc and Cbz groups. This allows for selective deprotection and functionalization without affecting other sensitive moieties. Unlike harsher traditional routes, this process operates under mild conditions (mostly -5°C to room temperature), significantly reducing energy consumption and minimizing the formation of degradation by-products, thereby enhancing the overall purity profile of the final intermediate.
Q: Is this synthesis route scalable for commercial production of reference standards?
A: Yes, the route is designed for scalability. The reagents used, such as di-tert-butyl dicarbonate and benzyl chloroformate, are commodity chemicals available in bulk. The reaction steps involve standard unit operations like extraction, washing, and column chromatography, which can be adapted for larger scale preparative HPLC or crystallization processes. The high yields reported in key steps, such as the quantitative deprotection, support its viability for generating sufficient quantities for global supply chains.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Docetaxel Impurity A Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality impurity standards play in the global pharmaceutical supply chain. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet your volume requirements with consistency. We adhere to stringent purity specifications and operate rigorous QC labs equipped with state-of-the-art analytical instrumentation to verify the identity and purity of every batch. Our commitment to technical excellence ensures that the Docetaxel Impurity A supplied meets the highest international regulatory standards.
We invite you to collaborate with us to optimize your supply chain for oncology drug development. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume needs. Please contact us to request specific COA data and route feasibility assessments for this and other complex pharmaceutical intermediates. Let us be your partner in achieving regulatory success and operational efficiency.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →