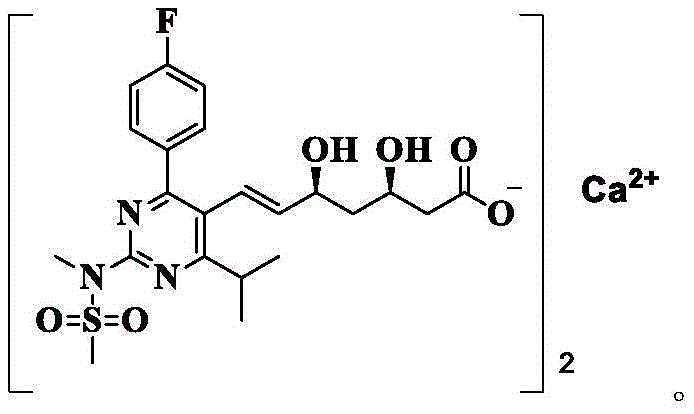
Optimizing Rosuvastatin Calcium Production: Advanced Purification of Key Intermediates
The pharmaceutical landscape for lipid-lowering agents continues to evolve, with Rosuvastatin Calcium standing as a cornerstone therapy for managing hypercholesterolemia. As detailed in patent CN109651259B, the synthesis of this potent HMG-CoA reductase inhibitor relies heavily on the quality of its precursors. The patent introduces a breakthrough purification methodology for the key intermediate, specifically the rosuvastatin methylamine salt, designated as Compound 4. This intermediate serves as the critical bridge between complex heterocyclic synthesis and the final active pharmaceutical ingredient. The structural complexity of Rosuvastatin Calcium, characterized by its polar methylsulfonyl amino group and dihydroxy heptanoic acid moiety, demands exceptional precision in intermediate processing to ensure bioavailability and therapeutic efficacy.
Traditional synthetic routes often struggle with the accumulation of stereoisomeric impurities that are notoriously difficult to separate in later stages. The innovation presented in this patent addresses this bottleneck by optimizing the crystallization parameters of the methylamine salt form. By strictly controlling solvent ratios and temperature gradients, manufacturers can achieve a significant reduction in diastereomeric impurities, thereby securing a robust supply chain for high-specification statin production. This technical advancement is not merely a laboratory curiosity but a scalable solution designed to meet the rigorous purity standards required by global regulatory bodies for reliable pharmaceutical intermediates supplier networks.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
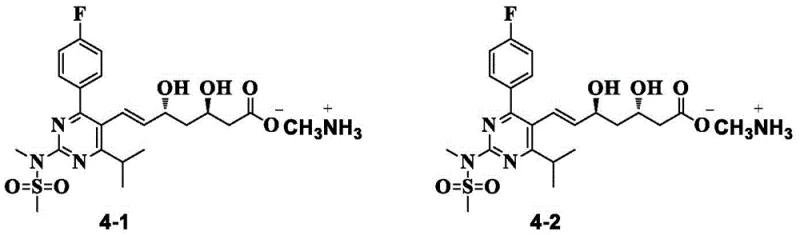
In the conventional synthesis of rosuvastatin intermediates, the formation of diastereoisomers presents a formidable challenge to process chemists. During the construction of the heptenoic acid side chain, particularly in the conversion of precursor compounds, a small but persistent amount of diastereoisomer is inevitably generated. These impurities, structurally identified as compounds 4-1 and 4-2, share nearly identical physical and chemical properties with the desired target molecule. In standard processing environments, these diastereoisomers are resistant to removal through typical washing or simple recrystallization techniques. Consequently, they persist through subsequent reaction steps, acting as carry-over contaminants that degrade the quality of the final API.
The presence of these isomers is particularly problematic because their structural similarity makes them invisible to many standard purification checks until the final stages, where remediation is most expensive. If left unchecked, these impurities can alter the pharmacokinetic profile of the drug, potentially leading to batch rejections and significant financial losses. The difficulty in purifying the intermediate at the methylamine salt stage has historically forced manufacturers to rely on costly chromatographic methods or accept lower overall yields, creating a bottleneck in the commercial scale-up of complex pharmaceutical intermediates.
The Novel Approach
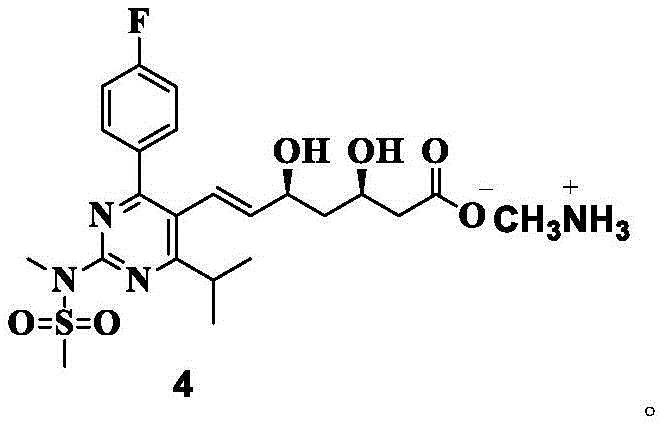
The patented methodology offers a paradigm shift by targeting the purification specifically at the Compound 4 methylamine salt stage. Instead of attempting to separate these stubborn isomers from the final acid or calcium salt forms, the process leverages the unique solubility characteristics of the methylamine salt. The innovation lies in the precise manipulation of a ternary solvent system. By dissolving the crude product in a specific ratio of alcohol and water, followed by the controlled addition of an ether solvent, the process creates a thermodynamic environment where the target compound crystallizes preferentially while the diastereoisomers remain in the mother liquor.
This approach effectively breaks the cycle of impurity transfer. The use of ethanol as the primary alcohol solvent and methyl tert-butyl ether (MTBE) as the anti-solvent provides a sharp solubility differential. The process operates within a defined temperature window, typically heating to 45-50°C to ensure complete dissolution before cooling to 0-10°C for crystallization. This thermal cycling, combined with the specific solvent polarity, ensures that the resulting crystals are of exceptionally high purity, with diastereomer content reduced to negligible levels (often below 0.05%). This represents a significant leap forward in cost reduction in pharmaceutical intermediates manufacturing by eliminating the need for downstream corrective actions.
Mechanistic Insights into Solvent-Mediated Crystallization
To fully appreciate the efficacy of this purification strategy, one must understand the interplay between the solvent system and the molecular lattice of the methylamine salt. The crude Compound 4 contains the target molecule alongside its diastereomeric counterparts. In a pure alcoholic environment, the solubility of both the target and the impurities may be too similar to allow for effective fractionation. However, the introduction of water modifies the dielectric constant of the medium, influencing the hydrogen bonding network around the polar hydroxyl and carboxylate groups of the molecule. The subsequent addition of the ether solvent, which is less polar and acts as an anti-solvent, reduces the overall solvation power of the mixture.
This reduction in solvation power forces the solute out of the solution. Crucially, the crystal lattice energy of the pure enantiomer (Compound 4) is distinct from that of the diastereomeric mixture. Under the optimized conditions described in the patent, the nucleation and growth kinetics favor the formation of the pure Compound 4 crystal lattice. The impurities, being structurally mismatched, are excluded from the growing crystal face and remain dissolved in the supernatant. This phenomenon, known as 'purification by crystallization,' is driven by the specific mass ratios of ethanol, water, and MTBE, ensuring that the saturation point is reached selectively for the desired isomer.
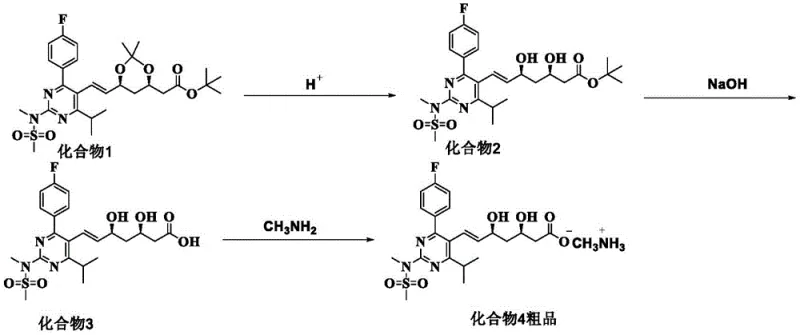
Furthermore, the synthesis of the crude Compound 4 itself involves a delicate sequence of acid-base reactions that set the stage for this final purification. As illustrated in the reaction scheme, the process begins with the deprotection of a ketal precursor (Compound 1) using protonic acid to yield the diol (Compound 2). This is followed by saponification with sodium hydroxide to form the sodium salt (Compound 3), which is then acidified and treated with methylamine. Each of these steps must be carefully controlled to minimize the initial formation of diastereomers, but the crystallization step acts as the final 'polishing' gatekeeper. Understanding this mechanistic flow is essential for any reliable pharmaceutical intermediates supplier aiming to replicate these results at scale.
How to Synthesize Rosuvastatin Methylamine Salt Efficiently
The synthesis and subsequent purification of Rosuvastatin Methylamine Salt require a disciplined approach to reaction conditions and solvent management. The patent outlines a robust protocol that transforms a crude mixture containing significant impurities into a high-purity intermediate suitable for final API synthesis. The process begins with the preparation of the crude salt via acidification and salt formation, followed immediately by the critical recrystallization step. Operators must pay close attention to the temperature ramps and the order of solvent addition, as these variables dictate the crystal habit and purity profile. For a detailed breakdown of the operational parameters, please refer to the standardized guide below.
- Dissolve the crude compound 4 in a mixture of alcohol solvent (preferably ethanol) and water, heating to 45-50°C.
- Add an ether solvent (preferably methyl tert-butyl ether) to the heated solution while maintaining temperature.
- Cool the mixture to 0-10°C to induce crystallization, then filter and dry to obtain high-purity product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this purification technology translates directly into operational resilience and cost efficiency. The traditional reliance on complex separation techniques often introduces variability in lead times and inflates the cost of goods sold. By implementing this streamlined crystallization process, manufacturers can stabilize their production schedules and reduce the dependency on expensive consumables. The ability to consistently produce high-purity intermediates minimizes the risk of batch failures, which is a critical factor in maintaining continuous supply lines for high-volume statin production.
- Cost Reduction in Manufacturing: The elimination of complex chromatographic purification steps results in substantial cost savings. Chromatography is capital-intensive, requiring specialized columns, large volumes of high-grade solvents, and significant labor for operation and column regeneration. By shifting to a crystallization-based purification, the process utilizes standard reactor vessels and common solvents like ethanol and MTBE. This simplification drastically lowers the variable costs per kilogram. Furthermore, the high recovery yield associated with this crystallization method ensures that raw material utilization is maximized, preventing the financial loss associated with discarding impure fractions.
- Enhanced Supply Chain Reliability: Supply chain continuity is often threatened by the inability to meet strict purity specifications, leading to quarantine and reprocessing delays. This purification method significantly mitigates that risk by ensuring that the intermediate meets high HPLC purity standards (>99.8%) right out of the crystallizer. When the intermediate quality is guaranteed, downstream synthesis steps proceed without interruption or unexpected deviations caused by impurity carryover. This predictability allows for tighter inventory management and more accurate delivery commitments to downstream API manufacturers, reinforcing the reliability of the supply chain.
- Scalability and Environmental Compliance: From an environmental and safety perspective, this process offers distinct advantages. The solvents employed (ethanol, water, MTBE) are well-understood, widely available, and easier to recover and recycle compared to the exotic solvent mixtures often required for chromatography. The simplicity of the unit operations—dissolution, heating, cooling, and filtration—makes the process inherently scalable from pilot plant to multi-ton commercial production. Additionally, reducing the solvent load and eliminating silica gel waste from chromatography columns aligns with green chemistry principles, helping manufacturers meet increasingly stringent environmental regulations and sustainability goals.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this purification technology. These insights are derived directly from the experimental data and technical specifications provided in the patent documentation, offering clarity on how this method resolves historical bottlenecks in rosuvastatin synthesis.
Q: Why is the removal of diastereoisomers critical in rosuvastatin synthesis?
A: Diastereoisomers (structures 4-1 and 4-2) possess physical properties very similar to the target compound 4. If not removed at the intermediate stage, they transfer through subsequent synthesis steps, severely compromising the purity and safety profile of the final rosuvastatin calcium API.
Q: What solvents are utilized in this novel purification method?
A: The process employs a ternary solvent system comprising an alcohol (optimally ethanol), water, and an ether solvent (optimally methyl tert-butyl ether). This specific combination maximizes the solubility difference between the target product and impurities.
Q: How does this method impact commercial scalability?
A: By replacing complex chromatographic separations with a straightforward crystallization protocol, the method significantly simplifies operations. It uses common, cost-effective solvents and standard temperature controls, making it highly suitable for multi-ton commercial manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Rosuvastatin Methylamine Salt Supplier
At NINGBO INNO PHARMCHEM, we recognize that the purity of intermediates defines the quality of the final medicine. Our technical team has extensively analyzed the pathways described in CN109651259B and possesses the expertise to implement this advanced crystallization technology at an industrial scale. We bring extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from lab bench to manufacturing floor is seamless. Our facilities are equipped with rigorous QC labs and adhere to stringent purity specifications, guaranteeing that every batch of Rosuvastatin Methylamine Salt we deliver meets the exacting standards required for global pharmaceutical registration.
We invite you to collaborate with us to optimize your supply chain for statin production. By leveraging our technical capabilities, you can secure a stable source of high-quality intermediates that drive down your overall manufacturing costs. Please contact our technical procurement team to request a Customized Cost-Saving Analysis. We are ready to provide specific COA data and route feasibility assessments tailored to your production volume requirements, ensuring that your project moves forward with confidence and efficiency.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →